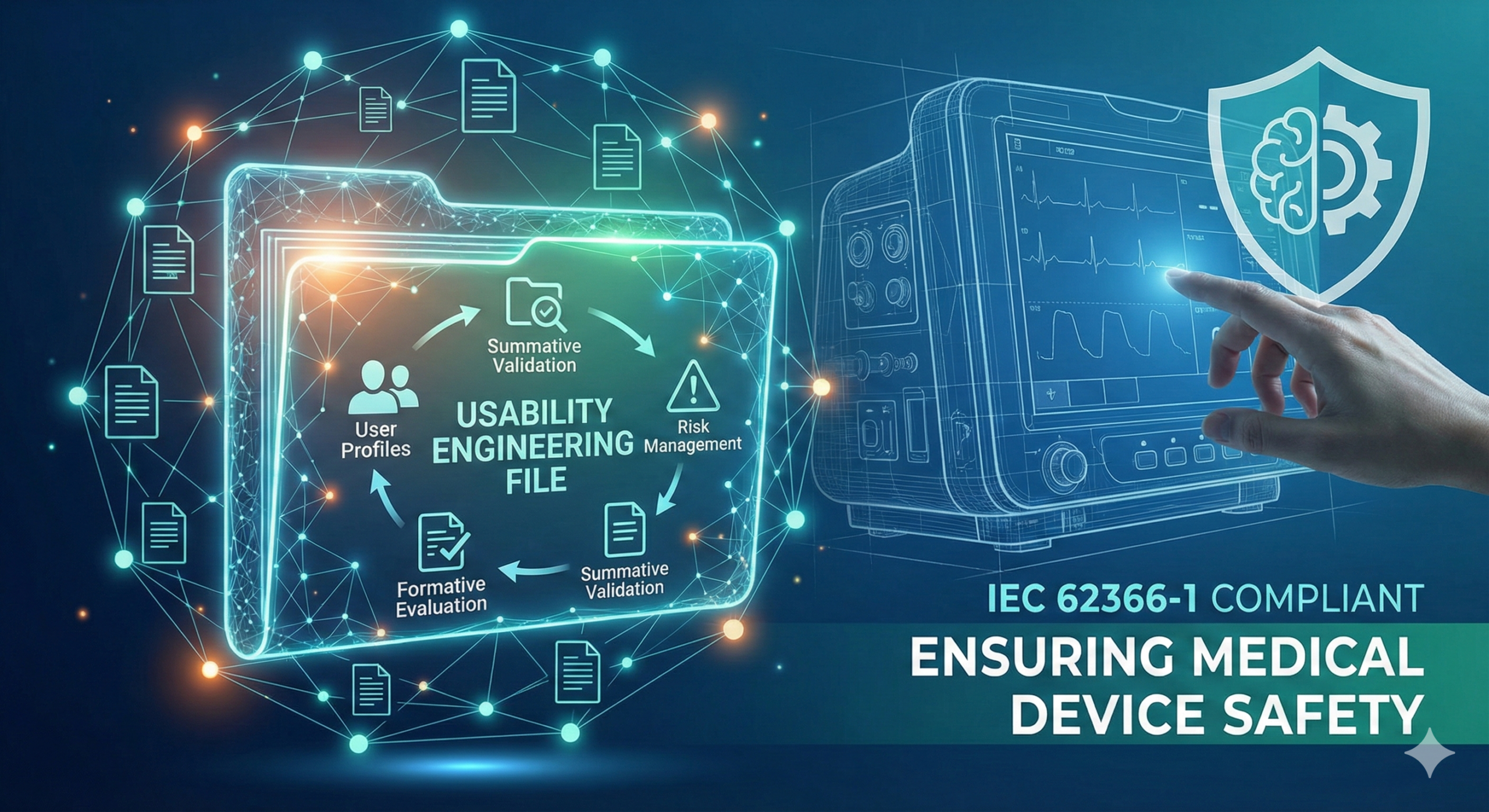
What is a Usability Engineering File?

The term “Usability Engineering File” (UEF) may sound unfamiliar to many people. To understand this specialized terminology, we must first explain the “Usability Engineering Process” in medical device regulation.
The term “Usability Engineering File” (UEF) may sound unfamiliar to many people. To understand this specialized terminology, we must first explain the…
The Role of Quality Standards in Medical Device Regulation
Medical device manufacturers are required to establish and maintain high quality standards. These requirements are established by international standards such as IEC 62366-1 (Usability Engineering of Medical Devices) and ISO 13485 (Quality Management Systems for Medical Devices). Each organization must adapt these standards to its own development process and implement them consistently across the entire organization.
The Essence of the Usability Engineering File
A Usability Engineering File is a documentation system that consolidates the activities conducted during the Usability Engineering Process, analytical results, verification data, risk assessments, and the rationale for design decisions. It serves as a comprehensive index indicating where various deliverables required by regulatory requirements and international standards are located and in what format they are stored.
The UEF is not limited to physical binders or folders; it is now implemented in various formats including digital systems, project management tools, and document management platforms. Fundamentally, it functions as an information mapping tool that connects regulatory requirements with actual evidence materials, or more precisely, as a traceability system.
The Relationship Between the UEF and Regulatory Requirements
Standards such as IEC 62366-1 explicitly require the implementation and documentation of the Usability Engineering Process for medical devices. This process proceeds through stages, beginning with the analysis of initial user needs, progressing through user interface design, formative evaluation, summative evaluation, and finally integration with risk management.
For example, to meet requirements that ensure safety for specific user populations, multiple forms of evidence are necessary. User task analysis results, records of usability testing activities, test cases and their outcomes, training materials provided to users, and documentation of design changes—all of these deliverables are managed centrally in the UEF. The UEF clearly indicates where these documents are located and how they correspond to specific regulatory requirements, thereby ensuring traceability during regulatory inspections and submissions for approval.
The Evolution of UEF Implementation Methods
The implementation of UEF in medical device development has evolved over time. The traditional approach involved completing the Usability Engineering Process first and then organizing and indexing the deliverables in the UEF retrospectively. While this method offers certainty, it presents a significant drawback: the development team cannot establish a clear visibility into regulatory compliance until later stages of development, creating a risk that additional work may be required as the project progresses.
Current best practices recommend a concurrent approach in which the UEF is constructed incrementally from project initiation. In other words, deliverables from Usability Engineering activities are recorded and indexed in real-time as the work progresses. Test plans, draft usability test reports, risk analysis tables, and successive versions of design specifications—intermediate deliverables from each stage of the development process—are reflected in the UEF.
This approach offers multiple advantages. First, by regularly consulting the UEF, the development team can monitor progress toward regulatory compliance and identify missing evidence or activities requiring additional implementation at an early stage. Second, because the rationale for design decisions is recorded in real-time, confusion and inconsistencies in later stages can be minimized. Third, the preparation of reports for quality assurance departments and regulatory authorities becomes more efficient and accurate.
Relevance to FDA Regulation
The FDA‘s “Human Factors Guidance for Medical Devices” explicitly requires documentation and record retention at each stage of the Usability Engineering Process, from planning through summative evaluation. The UEF functions as a practical tool for meeting these FDA requirements. Notably, in post-market surveillance and recall responses, usability engineering file records of design rationale and usability test results constitute critical evidence.
Similarly, under the EU’s MDR (Medical Device Regulation) and IVDR (In Vitro Diagnostic Regulation), technical file requirements mandate the presentation of evidence regarding usability and the rationale for design decisions. The UEF serves as the foundational system for meeting these regulatory requirements.
Typical Components of a UEF
The deliverables that a UEF should contain typically include the following elements. The Usability Engineering Plan documents the characteristics of target user populations, the intended use environment of the medical device, anticipated use errors and their associated risks, and an overview of planned Usability Engineering activities. Next, the UEF includes results from preliminary analysis such as user task analysis and use scenario analysis, prototypes of the user interface, and reports from formative evaluation activities, including multiple rounds of user testing. Additionally, the file should contain documentation demonstrating the integration of risk management processes with Usability Engineering activities, the plan and report for summative usability evaluation, and records of verification activities conducted on the final pre-market product.
The specific documents included in a UEF vary depending on the complexity of the medical device, the regulatory pathway (such as 510(k), PMA, or CE mark submission), and the structure of the company’s Quality Management System. The critical point is that all regulatory requirements are substantiated by actual deliverables contained within the UEF.
Continuous Improvement Through the UEF
The Usability Engineering Process continues beyond the market launch of a medical device. Feedback collected from users post-market, adverse event reports, and clinical data provide new insights into usability, which are incorporated into next-generation product development and software updates. Ideally, the UEF functions as a living documentation system that progressively incorporates post-market information.
Conclusion
The Usability Engineering File is a core tool for quality assurance and regulatory compliance in medical device development. It connects regulatory requirements with the actual deliverables from development activities, ensuring traceability and transparency. Properly constructing and maintaining the UEF at every stage—from the planning phase of the Usability Engineering Process through post-market management—is an essential requirement for developing and providing high-quality and safe medical devices. Effective utilization of the UEF is not merely a matter of regulatory compliance but rather a significant activity that contributes to improved user safety and satisfaction.
Related Articles
- What is Usability
- What is a Drug Master File (DMF)?
- The Importance of Use Environment and User Characteristics in Medical Device Usability Engineering
- Flow Rate and Volume Input Errors in Infusion Pumps: A Usability Engineering Perspective
- Aircraft Pilot Type Ratings and Medical Device Usability Engineering
- Scope of Application of Usability Engineering
{kind=link}
Comment