
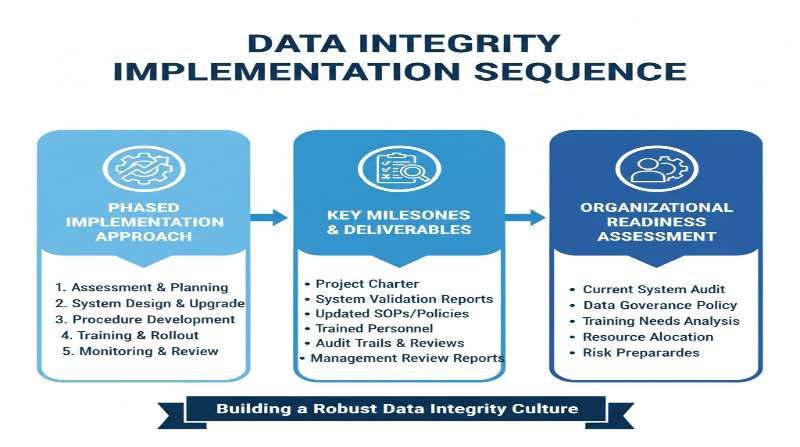
Data Integrity Implementation Sequence: A Comprehensive Guide for Pharmaceutical Organizations
Introduction
Data integrity has emerged as a critical compliance focus across global regulatory agencies. This guide outlines a systematic, risk-based approach to implementing robust data integrity controls in pharmaceutical manufacturing and quality operations. The sequence presented reflects current regulatory expectations from the FDA, EMA, MHRA, WHO, and PIC/S, emphasizing the foundational role of organizational culture and education in achieving sustainable data integrity compliance.
1. Education and Communication: Building the Foundation
The Critical Role of Education
Education represents the essential first step in establishing a comprehensive data integrity program. A well-documented phenomenon emerges when organizations initiate data integrity training: the number of identified violations initially increases. This increase does not reflect a deterioration in data quality; rather, it demonstrates enhanced awareness and detection capabilities among personnel. Previously unrecognized issues become visible as employees develop the competency to identify and report data integrity concerns.
This pattern mirrors similar dynamics in other domains. For instance, when medical representatives receive comprehensive adverse event training, the number of adverse event reports increases not because more events are occurring, but because recognition and reporting improve. Similarly, when traffic enforcement is increased, the number of documented speeding and parking violations rises due to enhanced detection rather than increased violation rates. These examples illustrate that discovering existing problems is the first step toward meaningful improvement.
Effective data integrity education programs should encompass several key elements. Personnel must understand the fundamental principles of data integrity, including the ALCOA+ framework (Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, and Available). According to current regulatory guidance, some organizations have adopted ALCOA++, which adds Traceable and other attributes to further strengthen data integrity controls. Training should be role-specific, ensuring that laboratory analysts, manufacturing operators, quality assurance personnel, and IT staff each understand their specific responsibilities and the data integrity risks inherent to their functions.
Furthermore, education must extend beyond initial training. Organizations should implement ongoing learning programs that address emerging regulatory expectations, new technologies, and lessons learned from both internal and external data integrity incidents. The FDA‘s 2024 draft guidance on data integrity for bioavailability and bioequivalence studies emphasizes that training personnel to detect data integrity issues is consistent with good manufacturing practice requirements.
Creating a Speak-Up Culture
Communication and organizational culture form the second foundational element of data integrity implementation. Recent regulatory requirements and international standards increasingly emphasize the critical importance of quality culture and psychological safety in preventing data integrity failures.
A common pattern in data integrity violations begins with a small error. When employees fear negative consequences, they may make a small misrepresentation to conceal the initial mistake. This small concealment often necessitates a larger deception, which in turn requires even greater fabrication. By the time the situation is discovered, the consequences have become severe, and the original issue has evolved into a systematic data integrity failure with serious regulatory implications.
Recognizing this dynamic, regulatory authorities and international standards now emphasize organizational cultures where errors can be reported freely without fear of punitive response. The FDA’s guidance on data integrity explicitly states that management with executive responsibility must create a quality culture where personnel understand that data integrity is an organizational core value and are encouraged to identify and promptly report data integrity issues. The MHRA‘s GxP Data Integrity Guidance (2018) similarly emphasizes that management should create a transparent, open work environment where personnel communicate failures freely so that corrective and preventive actions can be implemented.
Human error is inevitable in any complex operation. Organizations must recognize that mistakes will occur and establish systems that encourage early reporting. Rather than punishing individuals who report errors, companies should recognize and reward those who identify problems, report them promptly, and contribute to improvement initiatives. When errors are identified early, organizations can implement Corrective and Preventive Action (CAPA) systems to address root causes, revise procedures to prevent recurrence, and strengthen overall data integrity controls.
The PIC/S guidance on data integrity practices emphasizes that quality culture is fundamental to preventing intentional and unintentional data integrity failures. Organizations should evaluate their culture through observable behaviors, such as the willingness of personnel to raise concerns, the consistency of investigation practices, and leadership’s response to quality issues. Management must demonstrate through their actions that quality and data integrity take precedence over production pressures and schedule considerations.
2. Risk Identification and Mitigation
Understanding Process-Based Risk
Every pharmaceutical process contains inherent risks to data integrity. The second phase of implementation requires organizations to systematically identify where and how these risks exist within current standard operating procedures (SOPs) and to implement appropriate controls to mitigate them.
Consider a simplified manufacturing analogy to illustrate risk identification principles. Suppose a manufacturing process requires heating hamburger patties at 180°C for 3 minutes to ensure proper sterilization and food safety. If the temperature falls below 180°C or if the heating duration is less than 3 minutes, the patty center may not reach adequate temperature for proper sterilization, creating a food safety risk.
Breaking down this process reveals multiple potential failure modes. Temperature-related risks might include burner malfunction, inaccurate temperature measurement devices, or incorrect calibration of temperature gauges. Personnel might misread temperature displays or fail to verify that target temperature has been achieved before starting the timer. Duration-related risks include timer malfunction, incorrect timer settings, or premature removal of product before the required time has elapsed.
To address these risks, procedures must incorporate verification steps and controls. Equipment maintenance and calibration programs ensure measurement devices provide accurate readings. Independent verification requirements provide redundancy. Clear documentation standards ensure that any deviation from specified parameters is immediately visible and triggers appropriate investigation.
Applying Risk Assessment to Data Integrity
This same risk-based thinking applies directly to data integrity. Organizations should conduct systematic risk assessments of their data processes, examining each point where data is generated, recorded, processed, reviewed, and archived. The assessment should identify where data could be altered, deleted, or fabricated, whether intentionally or accidentally, and evaluate the potential impact of such failures on product quality and patient safety.
Risk assessment methodologies should follow established frameworks such as ICH Q9 (Quality Risk Management). Organizations should consider both technical controls (system-based safeguards) and procedural controls (human verification steps). Current guidance emphasizes that risk assessments should focus on business processes rather than solely on information technology functionality. Data flows should be mapped comprehensively, identifying all points where data moves between systems or transitions from one format to another.
The FDA’s data integrity guidance clarifies that CGMP regulations and guidance allow for flexible, risk-based strategies to prevent and detect data integrity issues. Organizations should implement meaningful and effective strategies to manage data integrity risks based on their process understanding and knowledge of technologies and business models. Not all data carries equal risk, and controls should be proportionate to the potential impact on product quality and patient safety.
Critical data points require more rigorous controls. For example, data directly supporting batch release decisions or stability studies warrant stronger protection than routine environmental monitoring records. However, organizations must ensure that all GMP-relevant data receives appropriate protection, as seemingly minor data can become critical during investigations or regulatory inspections.
Implementing Preventive Controls
Once risks are identified, organizations must implement controls to prevent data integrity failures. These controls typically span multiple categories:
Technical Controls: System configurations that prevent or detect inappropriate data manipulation, including access controls that limit system functions based on user roles, audit trails that record all data creation and modification activities, data backup and recovery systems that protect against data loss, and electronic signatures that ensure data is attributable and enforce workflow sequences.
Procedural Controls: Standard operating procedures that define acceptable practices for data generation, recording, and handling. These include witnessing requirements for critical operations, independent verification of calculations and transcriptions, defined processes for handling deviations and out-of-specification results, and clear documentation standards that ensure contemporaneous recording.
Physical Controls: Measures that protect physical records and restrict unauthorized access to data-generating equipment, such as controlled access to laboratories and manufacturing areas, secured storage for blank forms and completed records, locked computer terminals in restricted areas, and restricted access to system configuration utilities and administrative functions.
Organizations should document their risk assessments and the rationale for selected control strategies. This documentation demonstrates to regulatory agencies that data integrity controls are based on systematic evaluation rather than arbitrary choices. Regular review and updating of risk assessments ensures that controls remain appropriate as processes, technologies, and regulatory expectations evolve.
3. IT System Implementation and Enhancement
Addressing Technology-Mediated Data Integrity Risks
The third phase of data integrity implementation addresses technology systems used for data generation, processing, and storage. While technology offers powerful capabilities for ensuring data integrity, improperly configured or inadequately controlled systems can create significant vulnerabilities.
Risks of Common Systems
Spreadsheet applications such as Microsoft Excel present particular data integrity challenges. These versatile tools are widely used throughout pharmaceutical operations for calculations, data analysis, and record-keeping. However, standard spreadsheet configurations allow easy modification or deletion of data, whether intentional or accidental. Formula cells can be inadvertently overwritten with static values, eliminating the calculation logic. Data can be sorted incorrectly, creating associations between values that did not originally correspond. Entire rows or columns can be deleted, removing data without trace.
Without proper controls, spreadsheets fail to meet basic ALCOA+ principles. They typically do not create audit trails documenting who modified what data and when. Access control is often inadequate, allowing any user to modify any cell. Data backup may be inconsistent, depending on individual user practices rather than systematic procedures. The FDA and other regulatory agencies have issued numerous warning letters citing inadequate controls over spreadsheet-based systems.
Simple laboratory instruments, including electronic balances, pH meters, UV-visible spectrophotometers, and similar devices, present similar concerns. Many instruments, particularly older models, lack basic security features. They may not require user login, allowing any person to operate the equipment without identification. They may lack audit trail functionality, leaving no record of when measurements were taken, by whom, or whether data was deleted or modified. These deficiencies make it impossible to ensure data is attributable, contemporaneous, and original.
Implementing Appropriate System Controls
Organizations must evaluate their computerized systems against current data integrity requirements and implement necessary enhancements. The FDA’s guidance on data integrity clarifies that systems should be designed to maximize data integrity from the outset. For existing systems, organizations should implement controls through a combination of technical configuration and procedural safeguards.
For spreadsheet-based applications, organizations have several options. In some cases, spreadsheets can be replaced with validated software systems designed specifically for their intended purpose, such as laboratory information management systems (LIMS), electronic laboratory notebooks (ELN), or manufacturing execution systems (MES). When spreadsheets must be used, organizations should implement controls including: locked templates that prevent modification of formulas and structure; access controls limiting who can modify files; version control systems tracking all changes; regular data backups with integrity verification; and independent review of critical spreadsheets before use.
Some organizations implement specialized software that adds audit trail and access control capabilities to spreadsheet applications. These systems create a controlled environment around the spreadsheet, logging all access and modifications while restricting certain functions. This approach can be cost-effective for organizations with extensive spreadsheet use.
For laboratory instrumentation, organizations should prioritize replacing non-compliant systems with modern instruments that include built-in data integrity features. New instruments should be specified to include: user authentication requiring login before operation; comprehensive audit trails recording all operations and data; secure data transfer eliminating transcription steps; and tamper-evident design preventing unauthorized access to data storage.
When immediate replacement is not feasible, organizations must implement compensating controls through procedures, such as witnessed operations where a second person verifies and signs for critical measurements, contemporaneous logbook documentation of instrument use and results, regular verification of instrument data against recorded values, and physical security preventing unauthorized instrument access.
The Role of Electronic Systems in Data Integrity Assurance
Advanced electronic systems, properly implemented and maintained, provide superior data integrity assurance compared to paper-based systems or simple electronic devices. Modern laboratory and manufacturing systems can automatically enforce workflow sequences, prevent backdating of records, create comprehensive audit trails, and restrict system access based on user roles and training status.
However, organizations must recognize that technology alone does not ensure data integrity. Electronic systems require proper validation to ensure they function as intended and continue to meet requirements throughout their operational life. The FDA’s 21 CFR Part 11 establishes requirements for electronic records and electronic signatures, emphasizing that electronic systems must have appropriate controls to ensure authenticity, integrity, and confidentiality of records. The EU Annex 11 provides similar requirements for computerized systems.
Organizations implementing electronic systems should follow systematic approaches including: comprehensive user requirements specifying data integrity needs; risk-based validation demonstrating the system meets requirements; configuration management preventing unauthorized changes; routine monitoring verifying continued compliance; and periodic review ensuring systems remain appropriate for their intended use.
The PIC/S guidance emphasizes that organizations should understand their systems’ capabilities and limitations, including data lifecycle management, audit trail functionality, and security features. Personnel should be trained not only in routine system operation but also in recognizing and reporting potential data integrity issues within electronic systems.
4. Continuous Monitoring and Improvement
Sustaining Data Integrity Over Time
Data integrity is not achieved through a single implementation project but requires ongoing attention and continuous improvement. Organizations should establish monitoring programs to verify that controls remain effective and to identify emerging risks as processes, technologies, and regulatory expectations evolve.
Monitoring activities should include routine review of audit trails, examining patterns that might indicate circumvention of controls or system misuse; periodic data integrity assessments across different areas and processes; self-inspection programs specifically targeting data integrity vulnerabilities; review of deviations and CAPAs to identify systemic data integrity weaknesses; and trending of data integrity metrics to detect declining performance before serious issues arise.
Organizations should also maintain awareness of regulatory enforcement trends and emerging expectations. Regulatory agencies regularly publish warning letters, guidance documents, and inspection findings that provide valuable insights into current areas of concern. Participation in industry conferences, professional organizations, and collaborative initiatives helps organizations stay current with evolving best practices.
Current Regulatory Landscape and Future Directions
Key Regulatory Documents and Expectations
As of 2025-2026, pharmaceutical manufacturers should be familiar with several key regulatory documents addressing data integrity:
FDA Guidance: The FDA’s “Data Integrity and Compliance With Drug CGMP: Questions and Answers” (final guidance) provides comprehensive clarification of data integrity expectations within current good manufacturing practice. The April 2024 draft guidance “Data Integrity for In Vivo Bioavailability and Bioequivalence Studies” extends these principles to clinical and bioanalytical testing sites, emphasizing that sponsors and testing sites must implement quality management systems ensuring data integrity throughout the study lifecycle.
MHRA Guidance: The UK Medicines and Healthcare products Regulatory Agency’s “GXP Data Integrity Guidance and Definitions” (Revision 1, March 2018) remains a foundational document, providing detailed discussion of ALCOA+ principles and their application to both paper-based and electronic systems.
PIC/S Guidance: The Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme’s “Good Practices for Data Management and Integrity in Regulated GMP/GDP Environments” (PI 041-1, 2021) provides internationally harmonized expectations recognized by participating regulatory authorities worldwide.
WHO Guidelines: The World Health Organization’s Technical Report Series guidelines on data integrity provide global perspective and are particularly relevant for manufacturers operating in multiple markets.
EU Regulations: The European Union’s EudraLex Volume 4, particularly Annex 11 on Computerized Systems and ongoing revisions to various chapters, establishes binding requirements for EU-regulated operations. Recent developments include proposed incorporation of ALCOA++ principles explicitly in GMP regulations.
Emerging Trends and Focus Areas
Regulatory agencies continue to identify data integrity as a leading cause of warning letters, Form 483 observations, and import alerts. Common themes in recent enforcement actions include inadequate audit trails or failure to review audit trails, insufficient access controls allowing unauthorized data modification, failure to investigate anomalies and data integrity lapses, inadequate controls over computerized systems, and gaps between documented procedures and actual practices.
Looking forward, regulatory focus is expanding to include suppliers and contract organizations, emphasizing sponsor oversight obligations; data integrity in advanced therapy medicinal products and novel modalities; application of data integrity principles to real-world evidence and decentralized trials; and data integrity considerations in artificial intelligence and machine learning applications.
The FDA’s Quality Management Maturity (QMM) program, currently in prototype evaluation phase, is expected to incorporate data integrity as a key element of manufacturing quality system assessment. Organizations should consider data integrity implementation not as isolated compliance activity but as integral to overall quality management maturity.
Conclusion
Implementing robust data integrity controls requires a systematic, phased approach that begins with education and culture, progresses through risk assessment and mitigation, and culminates in appropriate technology implementation and continuous monitoring. Organizations that follow this sequence, with genuine commitment from executive leadership and engagement from all personnel, can achieve sustainable data integrity compliance that supports both regulatory requirements and the fundamental goal of ensuring pharmaceutical product quality and patient safety.
The data integrity journey is ongoing, requiring sustained attention, resources, and leadership commitment. However, organizations that embrace data integrity as a core value rather than merely a compliance obligation will find that these efforts enhance operational excellence, reduce risk, and strengthen confidence among regulators, customers, and ultimately the patients who depend on pharmaceutical products.
Summary Table: Data Integrity Implementation Sequence
| Phase | Key Activities | Expected Outcomes | Regulatory Basis |
| 1. Education & Communication | • ALCOA+ training for all personnel• Role-specific data integrity education• Speak-up culture development• Leadership commitment demonstration | • Increased awareness and detection• Enhanced error reporting• Psychological safety for quality concerns• Foundation for sustainable change | • FDA Draft Guidance (2024)• MHRA GxP Data Integrity Guidance• PIC/S PI 041-1• ICH Q10 |
| 2. Risk Identification & Mitigation | • Process-based risk assessments• Data flow mapping• Critical data point identification• Control strategy development• Procedural enhancement | • Documented understanding of risks• Prioritized control implementation• Reduced vulnerability to data integrity failures• Audit-ready risk rationale | • ICH Q9• FDA CGMP Data Integrity Q&A• PIC/S Data Integrity Guidance• MHRA Risk-based approaches |
| 3. IT System Implementation | • System capability assessment• Non-compliant system remediation• Enhanced audit trail implementation• Access control configuration• System validation | • Technology-enabled controls• Automated safeguards• Comprehensive audit trails• Reduced reliance on manual verification | • 21 CFR Part 11• EU Annex 11• PIC/S PI 011-3• GAMP 5 |
| 4. Continuous Monitoring | • Routine audit trail review• Data integrity self-inspections• CAPA trending and analysis• Regulatory intelligence monitoring• Periodic reassessment | • Sustained compliance• Early detection of emerging risks• Continuous improvement• Inspection readiness | • ICH Q10• FDA Quality Metrics• QMM Program Elements• International inspection trends |
Key Data Integrity Principles (ALCOA++)
| Principle | Definition | Practical Implementation | Common Vulnerabilities |
| Attributable | Data must be traceable to the individual who generated it | • User authentication systems• Electronic signatures• Unique user IDs | • Shared login credentials• Generic usernames• Unsigned records |
| Legible | Data must be readable and permanent throughout its lifecycle | • Permanent ink for records• High-resolution electronic displays• Protected storage media | • Fading printouts• Illegible handwriting• Degraded electronic media |
| Contemporaneous | Data recorded at the time the activity is performed | • Real-time data capture systems• Time-stamped entries• Workflow enforcement | • Batch documentation• Backdating entries• Pre-signed records |
| Original | First recording of data or certified true copy | • Source data preservation• Certified copy procedures• Metadata retention | • Undocumented transcriptions• Uncertified copies• Lost source data |
| Accurate | Data is correct and truthful | • Calibrated equipment• Verified calculations• Independent checks | • Transcription errors• Uncalibrated instruments• Calculation mistakes |
| Complete | All data collected is retained | • All trials documented• Failed analyses recorded• Repeat testing documented | • Cherry-picking data• Unreported failures• Deleted test runs |
| Consistent | Data recorded in correct sequence with reliable time stamps | • Synchronized system clocks• Sequential numbering• Logical chronology | • Timestamp manipulation• Out-of-sequence entries• Clock drift |
| Enduring | Data retained throughout required retention period | • Validated archive systems• Migration strategies• Redundant backups | • Media degradation• Format obsolescence• Lost backups |
| Available | Data must be readily retrievable for review | • Indexed storage systems• Documented retention locations• Format compatibility maintained | • Inaccessible archives• Obsolete formats• Lost documentation |
| Traceable | Complete audit trail throughout data lifecycle | • Comprehensive audit trails• Version control• Change documentation | • Disabled audit trails• Audit trail deletion• Unreviewed logs |
Related Articles
- The Birth of 21 CFR Part 11: A Historical Perspective
- Why Printouts Cannot Be Trusted: Understanding the Vulnerability of Paper-Based Document Management
- The Evolution of Electronic Records Regulations: From FDA Part 11’s Tumultuous Past to Today’s Global Harmonization
- Understanding Authenticity: A Core Requirement of Japanese ER/ES Guidelines
- Understanding Legibility: A Fundamental Requirement for Electronic Records
Related FDA QMSR Templates
Streamline your FDA QMSR compliance with our professionally crafted templates:



{kind=link}
Comment