
Practical Approaches to Human Error Prevention in Pharmaceutical Manufacturing: 4M Change Management and 3H Management

Introduction
During the morning meeting at a pharmaceutical manufacturing facility, the production manager began with a grave expression: “A weighing error occurred during the night shift yesterday. Fortunately, we discovered it before shipment, but if it had reached the market…” Such scenarios represent a reality that could occur at any pharmaceutical manufacturing facility.
During the morning meeting at a pharmaceutical manufacturing facility, the production manager began with a grave expression: “A weighing error occurre…
In pharmaceutical manufacturing, human errors are not merely quality issues. They harbor the potential to become serious incidents directly affecting patients’ health and sometimes their lives. If the amount of active pharmaceutical ingredient in a single tablet deviates from specifications, not only might the intended therapeutic effect fail to materialize, but risks of adverse effects from overdosing also emerge. Should microbial contamination occur during the manufacturing process of sterile preparations, there is potential for triggering severe infectious diseases.
Against this background, Good Manufacturing Practice (GMP) positions human error prevention as a core element of the quality assurance system. This article will explain two methodologies practiced in pharmaceutical manufacturing facilities—”4M Change Management” and “3H Management”—incorporating actual field experiences and case examples.
1. The Reality of Human Errors in Pharmaceutical Manufacturing Facilities
1.1 Why Errors Occur in Pharmaceutical Manufacturing Facilities
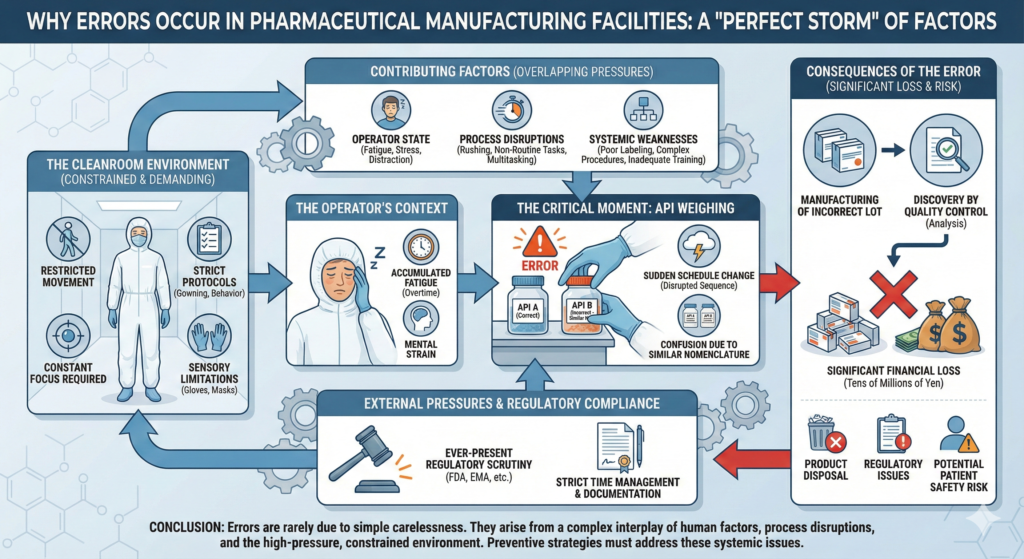
Let me introduce a real example from a pharmaceutical manufacturing facility. An experienced operator was weighing active pharmaceutical ingredients (APIs) in the same manner as always. However, that day, fatigue had accumulated from overtime work continuing from the previous day, and due to a sudden change in the production schedule, the work was being conducted in a sequence different from usual. As a result, APIs with similar names were confused, and incorrect raw materials were used. This error was discovered through analysis by the quality control department, but manufacturing of one lot had already been completed, leading to significant losses involving product disposal on the scale of tens of millions of yen.
As this case demonstrates, human errors in pharmaceutical manufacturing facilities are not caused solely by simple carelessness. They often occur when multiple factors—fatigue, sudden changes, confusion due to similar nomenclature—overlap. When such accidents occur, losses on the scale of tens of millions of yen from disposal of one product lot are not uncommon. Furthermore, factors specific to pharmaceutical manufacturing facilities, such as constrained work environments within cleanrooms, strict time management, complex manufacturing processes, and ever-present pressure regarding regulatory authority compliance, elevate error risks.
1.2 GMP’s Philosophy on Error Prevention
The 2025 revision of the Pharmaceuticals and Medical Devices Act (Pharmaceutical Affairs Act) has imposed stricter quality management systems on marketing authorization holders than ever before. Particularly noteworthy is the point that documentation and continuous improvement of human error prevention measures have been explicitly mandated. The 2025 revision, which was promulgated in May 2025, strengthened the responsibilities of marketing authorization holders, mandated the establishment of quality assurance managers and safety managers, and made GMP compliance more legally binding on manufacturing operators.
GMP’s fundamental philosophy is based on the premise that “humans inevitably make mistakes.” Therefore, rather than depending on individual attention, constructing mechanisms to prevent errors systematically is required. Specifically, thorough education and training of operators with periodic evaluation of their competence is necessary, as is the establishment of clear and comprehensible standard operating procedures (SOPs). When errors occur, root cause investigation must be conducted with certain implementation of corrective and preventive actions (CAPA). Additionally, all changes must be evaluated in advance and undergo appropriate approval processes.
2. Practical Implementation of 4M Change Management in Pharmaceutical Manufacturing Facilities
2.1 The Essence of the 4M Concept
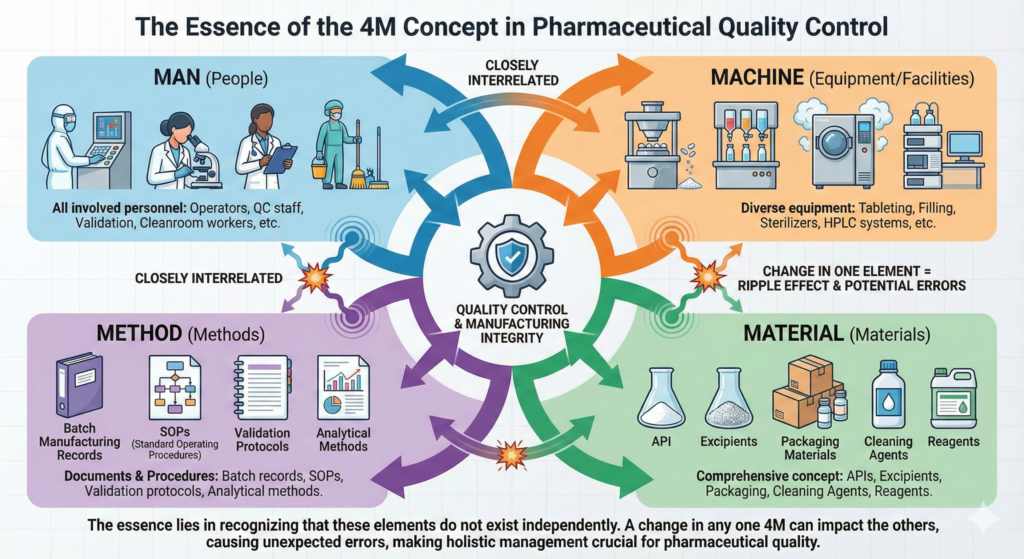
In quality control at pharmaceutical manufacturing facilities, the 4M concept is used routinely, but understanding its essence is important. 4M refers to the four critical elements affecting manufacturing: Man (people), Machine (equipment/facilities), Material (materials), and Method (methods).
“Man” in pharmaceutical manufacturing facilities refers not only to manufacturing operators but to all personnel involved in pharmaceutical manufacturing, including quality control personnel, validation personnel, and cleanroom workers. “Machine” encompasses diverse equipment such as tableting machines that form tablets, filling machines that fill injections, sterilizers that sterilize products, and HPLC (High Performance Liquid Chromatography) systems that analyze quality. “Material” is a comprehensive concept that includes not only direct materials such as APIs and excipients but also packaging materials, cleaning agents, and reagents. “Method” means all documents and procedures that regulate work methods, such as batch manufacturing records, SOPs, validation protocols, and analytical methods.
These four elements do not exist independently but are closely interrelated. When a change occurs in one element, the impact can ripple to other elements, potentially causing errors in unexpected places.
2.2 The Reality of Change Management Systems
In GMP, change control is a mandatory requirement. All changes must be evaluated for their impact on quality and receive appropriate approval. While this requirement may seem cumbersome, understanding its importance becomes clear when examining cases that have occurred at actual pharmaceutical manufacturing facilities.
At a certain pharmaceutical manufacturing facility, the punch and die of a tableting machine on a tablet manufacturing line became worn and required replacement. While seemingly a simple parts replacement, the quality assurance department conducted a careful impact assessment. Multifaceted examination was necessary, including the possibility that the new punch and die might change tablet hardness, potential impact on disintegration properties, consistency with already validated parameters, and impact on stability studies.
The change request first receives confirmation from the manufacturing department head, then approval from the QA department head, and finally from the quality assurance manager. While this process is time-consuming, having checks from different perspectives at each stage prevents oversights. After actual punch and die replacement, the initial lot is intensively managed, and only after confirming that all quality items are within specifications is the transition made to normal production.
2.3 Learning from a Case of API Supplier Change
Let us examine in detail an actual case of API supplier change. For the main product of a certain pharmaceutical company, from the perspective of supply stability and cost reduction, a switch from an overseas manufacturer to a domestic manufacturer was considered for the API used.
First, the quality control department initiated detailed analysis of the new API. In comparing impurity profiles, the types and amounts of impurities contained were precisely analyzed to confirm equivalence with the existing product. In confirming particle size distribution and crystal form, the impact of the API’s physical properties on the formulation process was evaluated. In analyzing residual solvents, it was confirmed that the types and amounts of solvents used were within specifications.
During this evaluation process, it was discovered that the new API had slightly larger particle diameter, potentially resulting in insufficient penetration of binding agent during the granulation process. Therefore, after consultation with the manufacturing department, optimization of granulation conditions was implemented. Specifically, by increasing the amount of binding agent by 5% from the conventional formulation and raising the supply air temperature of the fluid bed granulator by 5°C, appropriate granulation became possible.
Accompanying this change, two hours of classroom education were conducted for all operators, followed by practical training. The education explained in detail why the change was necessary, what points required attention, and how to respond if abnormalities occurred.
As validation, three lots were manufactured under new conditions, confirming that all quality items were within specifications. Furthermore, accelerated stability testing (40°C, 75% relative humidity for 6 months) and long-term stability testing (25°C, 60% relative humidity for 36 months) were initiated to confirm quality stability over time.
Through this series of efforts, while maintaining quality within specifications even after the change, stabilization of API procurement and significant cost reduction were achieved.
2.4 Comprehensive Management in Aseptic Filling Line Renewal
In injection manufacturing, ensuring sterility is the most important quality element. Through a case where an aging aseptic filling line was updated to the latest equipment, let us examine how Machine (equipment) changes affect the other three M elements.
The new aseptic filling machine improved filling accuracy from ±3% to ±1% compared to conventional machines, and filling speed doubled from 100 bottles/minute to 200 bottles/minute. Furthermore, by adopting an isolator system, it became possible to minimize contact opportunities between operators and products.
However, this equipment renewal did not simply end with machine replacement. First, regarding the human element, glove operation within the isolator required completely different techniques from conventional work in open systems. Therefore, a new operator certification system was introduced, conducting 40 hours of glove operation proficiency training. As confirmation of aseptic operation, media fill testing was conducted using media instead of actual product, proving that each worker could work aseptically.
Regarding materials, compatibility confirmation of rubber stoppers was necessary. As the thickness and speed of needles on the new filling machine changed, evaluation of impact on rubber stoppers was necessary, requiring optimization of silicone oil application amount. The method of supplying vials was also changed to match the new equipment.
Regarding methods, new SOPs were created for 15 documents, environmental monitoring plans were reviewed, and process control values were reset. These documents were refined into practical ones suited to actual field conditions through repeated revisions while performing actual work.
3. Practical Implementation of 3H Management in Pharmaceutical Manufacturing Facilities
3.1 The Importance of the 3H Perspective
Anyone working at pharmaceutical manufacturing facilities has experienced making mistakes they wouldn’t normally make when facing situations that are “different from usual.” 3H management systematically captures these “different from usual” situations. 3H stands for Hajimete (first time), Henkou (change), and Hisashiburi (after a long time), representing three situations where errors are likely to occur.
“First time” in pharmaceutical manufacturing facilities refers to situations of inexperience, such as when new employees begin working in cleanrooms, when manufacturing of a new product begins, when a new analytical method is introduced, or when responding to a regulatory authority inspection for the first time. “Change” includes changes in manufacturing scale, changes in API particle size specifications, changes in packaging specifications, and changes in manufacturing location through technology transfer. “After a long time” refers to work that has not been performed for an extended period, such as resumption of manufacturing seasonal products like hay fever medications, annual aseptic assurance confirmation (media fill), periodic disassembly and cleaning of equipment, and annual product quality review.
It is widely recognized in pharmaceutical industry quality control practice that error occurrence rates increase significantly in these non-routine states compared to routine work. At actual manufacturing sites, significant effects on error prevention have been achieved through special management of 3H states.
3.2 When 3H Risks Become Reality
Let me introduce an actual case. At a certain pharmaceutical manufacturing facility, a specific injection was to be manufactured for the first time in six months. This product had large demand fluctuations, and manufacturing intervals tended to be irregular due to inventory adjustments.
On the day of manufacturing, experienced operators began work, but several problems occurred in a chain reaction. First, the filling amount was out of specification. Upon investigating the cause, it was discovered that the filling machine’s set values had been entered incorrectly. Even more seriously, microbial contamination was detected in sterility testing. This was caused by overlooking a pH adjustment method that had been changed since the previous manufacturing run and a memory error regarding glove change frequency. To compound matters, errors were also found in product labels. Despite design changes, old version labels were used.
As a result, the manufactured batch of products had to be disposed of. In pharmaceutical manufacturing facilities, manufacturing one batch can cost tens of millions of yen, making such disposal a significant loss for the company. Furthermore, investigating causes and implementing corrective measures, and preparing for remanufacturing required two weeks, resulting in delayed supply to customers. This case clearly demonstrates how dangerous the 3H state of “after a long time” can be.
3.3 3H Management Practice in API Weighing Process
API weighing is one of the most important processes directly affecting product efficacy. Minor errors can affect patient therapeutic outcomes, and particularly for highly active pharmaceuticals and narcotics/psychotropics, meticulous care is required in handling. At a certain pharmaceutical manufacturing facility, comprehensive 3H management is implemented in this critical process.
When new employees perform API weighing for the first time, a staged certification system is adopted. Beginning with Level 1, weighing of general APIs starts. After performing 10 operations under supervisor supervision and meeting standards in all cases, independent work is permitted. At Level 2, handling of highly active APIs becomes possible, but this requires an additional 20 hours of specialized training. Level 3 qualification for weighing narcotics and psychotropics requires special certification by management.
In actual training, practice begins using placebo powder. Weighing is performed using the same procedures as with actual APIs, measuring and evaluating errors. Donning and doffing of protective equipment is also mastered while understanding contamination risks. Through this staged approach, operators gradually develop confidence in performing accurate work.
Countermeasures when changes occur are also important. For example, when weighing methods change, change notification cards are displayed at the entrance to the weighing room. Changes are highlighted in red text, and signature columns for confirmation ensure that everyone has recognized the change. Furthermore, for one week after the change, work is always conducted with a two-person system, and items reflecting change points are added to checklists.
For work after a long time, management through a skills matrix is effective. The system records the last execution date for each operator, automatically issuing alerts for operations not performed for three months or more. Relevant personnel must take a procedure confirmation test before work and perform a reading of key points before beginning work.
3.4 Annual Validation as a Special 3H Task
Annual validation of sterilizers is a典型example of “after a long time” work. It is special work performed only once per year, with complex procedures, and if it fails, it has potential to affect the entire manufacturing line.
At a certain pharmaceutical manufacturing facility, meticulous 3H management is implemented for this annual validation. Preparation begins one month before implementation, first reviewing the previous execution record in detail. Problems that occurred the previous year and improvement items are confirmed, along with a comprehensive check of specification changes and equipment updates that occurred in the interim.
In team composition, experienced and inexperienced personnel are intentionally paired. This simultaneously realizes technology transfer and introduction of new perspectives. Each member’s responsibility range is clarified, and who is responsible for what is determined in advance.
On the day of implementation, it begins with a morning briefing. Procedures are confirmed again, anticipated risks are shared, and emergency responses in case of unforeseen circumstances are also confirmed. In the work area, “Annual Validation in Progress” signage is posted to make surrounding personnel aware of the unusual state. Confirmation stamps are affixed at each checkpoint, visualizing work progress.
Particularly important is real-time recording. The idea of “recording later” is strictly prohibited, with records made on-site with each operation. Photographic documentation is conducted in parallel, ensuring a state that can be verified later.
In the verification phase, obtained results are compared with past data, carefully confirming absence of abnormal values. Finally, matters for next time are created, and reflection in SOPs is considered as necessary. Through such a systematic approach, even special tasks occurring once annually can be successfully completed with certainty.
4. Integrated Application of 4M Change Management and 3H Management in Pharmaceutical Manufacturing Facilities
4.1 When Two Management Methodologies Converge
While change management is mandated in the GMP world, combining it with 3H management enables deeper insights and more effective risk management. FDA (Food and Drug Administration) inspectors also frequently focus on human error countermeasures during changes. This is because regulatory authorities also recognize the importance of integrating the 4M perspective of change with the human cognitive perspective of 3H states that changes create.
4.2 Integrated Management Reality in New Drug Technology Transfer
Technology transfer to scale up new drugs born in research and development departments to commercial production scale represents one of the most complex projects in pharmaceutical manufacturing facilities. Through a technology transfer project actually conducted at a pharmaceutical company, let us examine the power of integrated application of 4M change management and 3H management.
This project began with 4M and 3H matrix analysis. For the Man (people) element, handover from development personnel to manufacturing personnel occurs, clearly creating a “first time” state. Machine (equipment) is a transition from laboratory-scale experimental equipment to commercial production equipment, corresponding to both “first time” and “change.” Material (materials) is a switch from reagent grade to GMP grade, constituting “change,” and Method (methods) is a formulation change from small scale to large scale, also classified as “change.”
This analysis clearly revealed that equipment and people elements had the highest risk. Therefore, the technology transfer team was composed of members selected from development, manufacturing, and quality assurance departments, with mandatory 3H state confirmation at weekly progress meetings.
Scale-up was not performed all at once but progressed gradually through 10 liters, 100 liters, 1000 liters, and finally 5000 liters. By confirming 4M change points at each stage and conducting 3H evaluation, problems could be discovered and addressed early.
Particularly important was Knowledge Transfer. Development personnel conducted two weeks of OJT at the manufacturing site, not merely teaching procedures but explaining in detail why those conditions were necessary and which parameters were important (Critical Process Parameters). Additionally, a troubleshooting guide was created including past failure cases, enabling manufacturing personnel in “first time” states to respond appropriately when facing problems.
In the validation strategy, video recordings were made of 3H work, enabling later verification. When deviations occurred, root cause analysis was conducted from both 4M factors and 3H factors perspectives.
As a result of this integrated approach, the technology transfer period was shortened by two months from the schedule, and success was achieved in manufacturing specification-compliant products from the first batch. Even more impressively, deviations in the first year of commercial production were reduced to zero. This is the result of fusing systematic approach through 4M change management with human-centered perspective through 3H management.
4.3 Integrated Management System in the Digital Era
In modern pharmaceutical manufacturing facilities, electronic Quality Management Systems (eQMS) are widely introduced. By incorporating 4M change management and 3H management into these systems, more effective management becomes possible.
In change management modules, 4M classification is automatically required upon application, and 3H states that each change triggers are simultaneously evaluated. The system searches for past similar changes and presents problems that occurred then and countermeasures taken. This can significantly reduce the risk of repeating the same failures.
Linkage with education and training modules is also important. 3H applicable personnel are automatically extracted, and necessary education is automatically assigned. For example, when a new analytical instrument is introduced, notifications for education reception are automatically sent to all analytical personnel who might use that instrument. Reception status and competence evaluation results are also recorded and can be immediately presented during inspections.
In risk assessment, occurrence probability is evaluated by number of 3H applicables (none is 1 point, 1 is 3 points, 2 or more is 5 points), severity is evaluated by 4M change impact degree (limited is 1 point, moderate is 3 points, extensive is 5 points), and detectability is evaluated by difficulty of detection in quality testing (easy is 1 point, normal is 3 points, difficult is 5 points). Response priorities and approval levels are automatically determined through risk scores calculated by multiplying these factors.
5. The Path to Introduction and Establishment in Pharmaceutical Manufacturing Facilities
5.1 GMP Culture as Foundation
GMP culture is already deeply rooted in pharmaceutical manufacturing facilities. This existing culture represents a significant advantage in introducing 4M change management and 3H management. This is because GMP’s three principles—”minimize human errors,” “prevent contamination and quality degradation,” and “design systems to assure high quality”—align precisely with what 4M change management and 3H management aim to achieve.
What is important is positioning these management methodologies not as new burdens but as means to strengthen existing GMP systems. At many pharmaceutical manufacturing facilities, systems for change management and education/training already exist. 4M change management and 3H management are tools that provide new perspectives and structure to these systems, making them more effective.
5.2 Value as Preparation for Inspections
Regulatory authority inspections are unavoidable checkpoints for pharmaceutical manufacturing facilities. Inspectors thoroughly verify change management system effectiveness, education/training appropriateness, and evidence of continuous improvement.
At a certain pharmaceutical manufacturing facility, there was experience of deep pursuit regarding change management during an FDA inspection. The inspector asked, “What risks did you anticipate with this change, and how did you address them?” At that time, by presenting records of 4M analysis and 3H evaluation, systematic risk evaluation and response could be clearly explained. The inspector evaluated it as a “Very systematic approach,” and there were no findings on this point.
At another inspection, questions were asked about education/training effectiveness. By explaining the staged certification system based on 3H management and the management system through skills matrices, comments were received that “This is a good example that should be referenced by other facilities.”
5.3 Education Holds the Key to Transformation
It is no exaggeration to say that success of 4M change management and 3H management in pharmaceutical manufacturing facilities depends on education and training. Education programs appropriate for each level are necessary, from new employees to managers.
For new employees, 16 hours are spent teaching GMP fundamentals along with 4M/3H concepts in new employee training. However, since classroom learning alone does not provide a sense of reality, cleanroom changing practice and simulated 3H experience training are incorporated. For example, by intentionally creating confusing situations and having them experience errors, understanding of why errors occur easily is fostered.
For site operators, 8 hours are spent at the time of assignment to understand 4M elements of their assigned processes. They concretely learn which 4M elements their work relates to and what changes might occur. Subsequently, 40 hours of OJT develop practical skills.
For leaders and supervisory personnel, upon promotion, 16 hours are spent teaching 4M change management planning methods. Ability is required not just to follow procedures but to anticipate changes and manage them appropriately. 3H management guidance methods are also learned, enabling them to identify subordinates’ 3H states and provide appropriate support.
For managers, annual updates on regulatory trends are conducted for 4 hours. Additionally, inspection response simulations are conducted to prepare for appropriate answers to inspector questions. Best practices from other facilities are also shared, leading to continuous improvement.
Particularly effective is error experience training. In weighing error experience, API containers with similar names are intentionally arranged, and situations are created that urge haste. Many participants make errors, but this enables them to realize the danger of 3H states. In aseptic operation simulation, bacterial culture media are manipulated in glove boxes, intentionally setting up situations with high contamination risk. When contamination is confirmed after culture, participants understand the difficulty and importance of aseptic operations through physical experience.
6. Effect Measurement and Continuous Improvement at Pharmaceutical Manufacturing Facilities
6.1 The Trajectory of Improvement Told by Numbers
In pharmaceutical manufacturing facilities, objectively evaluating the results of improvement activities is extremely important. At a certain pharmaceutical manufacturing facility, analysis of three years of data after introducing 4M change management and 3H management revealed interesting trends.
Examining deviation number trends, there was a temporary increase in the first year of introduction. While this appears to be an adverse trend, it was actually the result of heightened recognition of 3H states making risks that had previously been overlooked become manifest. From the second year onward, deviation numbers steadily decreased, with Critical (severe) level deviations in particular decreasing by 75% from 12 annually before introduction to 3 per year.
Analyzing in more detail, clear patterns were observed in human error occurrence rates by day of week. Errors concentrated on Mondays and Fridays, thought to be influenced by week-start startup and end-of-week fatigue. Errors also increased during shift-change handover times. Based on this discovery, measures were implemented to concentrate important work in the day-shift period from Tuesday through Thursday, achieving a further 20% reduction in error rates.
Out of Specification (OOS) rate is also an important indicator. Particularly noteworthy is that the proportion of OOS attributable to human error decreased from 45% before introduction to 15%. This led to cost reductions on the scale of hundreds of millions of yen annually. This is because when OOS occurs, cause investigation, retesting, and in some cases product disposal are necessary, requiring enormous costs and time.
6.2 The Power of Prevention Shown by Data
The pharmaceutical manufacturing facility’s data analysis team discovered possibilities for preventive management from accumulated data. For example, analyzing equipment trouble occurrence rates when new employees were assigned revealed that troubles occurring at three times the normal rate within two weeks of assignment. Detailed cause analysis revealed that this was primarily due to operational errors from insufficient education.
Based on this discovery, an equipment-specific certification system was introduced. New employees progressively acquire operation qualifications for equipment, with a mechanism where independent operation is permitted only after a minimum of 10 guided operations for each piece of equipment. After introduction of this system, equipment troubles caused by new employees decreased by 80%.
Seasonal variation analysis also showed interesting results. A clear tendency emerged for deviations related to temperature control to increase in summer and weighing errors due to static electricity to increase in winter. Based on these findings, season-specific SOPs were formulated. Summer SOPs specify strengthened air conditioning management and frequent temperature confirmation, while winter SOPs strengthen humidity control and static elimination countermeasures.
6.3 Best Practices Learned from Successful Companies
The case of major domestic pharmaceutical company A is particularly instructive. While this company manages approximately 500 changes annually, it succeeded in reducing deviations caused by changes by 83%, from 30 annually before introduction to 5. The key to success lay in optimization of risk-based approval processes. Rather than managing all changes at the same level, by varying approval levels and required verification depth according to risk, both efficiency and certainty were achieved.
Another pharmaceutical company B shortened startup period for new product introduction from 6 months to 3 months through strengthened 3H management and improved initial lot pass rate from 70% to 95%. Their success factor was enhancement of simulation training. By repeatedly practicing in virtual environments before actual manufacturing, a state that was no longer “first time” was created for actual manufacturing.
7. Latest Trends in the Pharmaceutical Industry and Future Outlook
7.1 Transformation Brought by Regulatory Environment Evolution
In 2025, the pharmaceutical industry is at a major turning point. With implementation of ICH Q12 (Pharmaceutical Product Lifecycle Management) progressing, the paradigm of change management is shifting. Previously, notification to regulatory authorities was required for even minor changes, but by agreeing in advance to Change Management Protocols (CMP), changes within a certain scope can now be implemented at company discretion.
While this appears to be regulatory relaxation, it actually means that more advanced change management capabilities are required of companies. In this new regulatory environment, 4M change management and 3H management are becoming important tools for companies to autonomously assure quality.
Data integrity requirements are also becoming stricter year by year. ALCOA+ principles (Attributable, Legible, Contemporaneous, Original, Accurate, plus Complete, Consistent, Enduring, Available) have become basic requirements demanded of all records. Records of 4M change management and 3H management are no exception, with electronic systems requiring audit trail integrity, strict access permission management, and reliable backup and recovery systems. The framework has been further extended to ALCOA++, adding Traceable, adding more stringent requirements.
7.2 New Possibilities Brought by Pharma 4.0
The wave of the Fourth Industrial Revolution is also reaching the pharmaceutical industry, with attention gathering on the concept of “Pharma 4.0.” In particular, continuous manufacturing represents an approach completely different from conventional batch production, requiring new perspectives on 4M change management and 3H management.
In continuous production lines, real-time quality monitoring through PAT (Process Analytical Technology) and immediate detection and response to deviations are becoming increasingly possible. Continuous monitoring of 4M parameters has also become technically feasible, with construction of systems that do not miss even minor changes progressing. Development of systems supporting 3H state risk assessment using machine learning algorithms is also advancing, with future automatic suggestion of optimal operating conditions being anticipated. However, it should be noted that many of these technologies are still in implementation and verification stages, and complete automation has not yet been achieved.
Digital twin technology is also bringing about transformative change. Efforts to construct digital models of manufacturing lines in virtual space and simulate 4M change impacts in advance have begun. Some advanced companies are constructing virtual training environments for 3H work, realizing education and training in risk-free environments. Digital twins are also beginning to be utilized as effective tools when comparing multiple change proposals and examining the balance between cost and quality.
7.3 Globalizing Quality Assurance
Many modern pharmaceutical companies have manufacturing bases worldwide and conduct business through networks with CMOs (Contract Manufacturing Organizations) and CDMOs (Contract Development and Manufacturing Organizations). In this global manufacturing network, standardization of 4M change management and 3H management has become an extremely important issue.
A Japanese global pharmaceutical company constructed unified 4M/3H management standards at 30 bases worldwide. A multilingual electronic system was introduced, and mechanisms were created for horizontal deployment of best practices. Similar management is also required of contracted CMOs/CDMOs, with strengthened 3H management during technology transfer and incorporation into regular audit verification items.
Application to biopharmaceutical manufacturing is also advancing. Cell culture processes involve handling living organisms and thus have greater variability, making 4M management more complex. The importance of aseptic operations is also significantly higher, making strengthened 3H management indispensable. Scale-up risks also differ from conventional small molecule pharmaceuticals, requiring new approaches.
Conclusion
The story that began with the weighing error discussed at the pharmaceutical manufacturing facility morning meeting presents one answer to the universal challenge of human error prevention. 4M change management and 3H management are not merely management methodologies but rather a philosophy for protecting patient safety and a crystallization of practical wisdom.
As we have seen, success at pharmaceutical manufacturing facilities is not accidental. It is realized through integration of systematic approach through 4M change management and human-centered perspective through 3H management on the firm foundation of GMP. Through risk-based approaches, appropriate management levels are set according to impact degree. Through data-driven improvement, deviation trends are analyzed and preventive measures are implemented. Response to regulatory requirements is viewed not as mere compliance but as an opportunity for quality improvement. Above all, technological innovation is actively utilized to achieve digitalization-driven efficiency and accuracy improvements. Most importantly, a global perspective is maintained, promoting adaptation to international standards and standardization.
In the future, pharmaceutical manufacturing will enter a further transformation period with continuous production, personalized medicine, and other developments. Manufacturing processes will become more complex, product portfolios will diversify, and regulatory requirements will become more sophisticated. In such environments, the importance of 4M change management and 3H management will only increase.
However, no matter how much technology advances, humans will always be at the center of pharmaceutical manufacturing. And humans are beings who make mistakes. Humbly accepting this fact and continuing to construct mechanisms to systematically prevent errors is the mission of those of us engaged in the pharmaceutical industry.
Beyond a single tablet or a single injection lies a patient who needs it. To protect that patient’s life and health, through 4M change management and 3H management, we continue to stand today as guardians of quality. It is not flashy work, but it is work that should be continued reliably, steadily, and sincerely.
To quality assurance personnel, manufacturing managers, and regulatory compliance managers at pharmaceutical manufacturing facilities, I sincerely hope you will adapt the concepts and methodologies introduced in this article to your company’s situation. While perfect systems do not exist, approaching perfection through continuous improvement is possible. Believing that the accumulation of those efforts will support tomorrow’s healthcare and lead to patient smiles.
References
Ministry of Health, Labour and Welfare “Ministerial Ordinance on Standards for Manufacturing Control and Quality Control for Drugs and Quasi-drugs” (GMP Ordinance)
FDA “Guidance for Industry: Process Validation – General Principles and Practices”
ICH Q12 “Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management”
PIC/S GMP Guide Annex 15: Qualification and Validation
ISPE “Pharma 4.0™ Operating Model – Enabling Pharma 4.0 in Your Organization”
Pharmaceuticals and Medical Devices Agency (PMDA) “GMP Case Studies”
Japan Pharmaceutical Manufacturers Association “Human Error Countermeasures in Pharmaceutical Manufacturing”
EMA “GMP Annex 11: Computerised Systems”
WHO “Technical Report Series No. 996, Annex 5: Guidance on Good Data and Record Management Practices”
MHRA “GxP Data Integrity Guidance and Definitions”
Related FDA QMSR Templates
Streamline your FDA QMSR compliance with our professionally crafted templates:

Related Articles
- Mechanisms to Prevent Human Error: 4M Change Management and 3H Management
- Understanding Self-Inspection in Pharmaceutical Quality Management: A Comparative Analysis of Global Regulatory Approaches
- Issuance of Administrative Notice on Manufacturing and Quality Management Systems for Pharmaceutical Manufacturers
- 4M Change Management in Medical Device Manufacturing Processes
- Preventive Action as Risk Management: Understanding CAPA in Pharmaceutical and Medical Device Industries
- Quality Risk Management in the Pharmaceutical Industry
{kind=link}