The Latest Timeline and Implementation Status of the Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR)

Overview of the Regulatory Framework
The European Commission publishes a Rolling Plan that outlines the implementation timeline and status of the Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR), which represent a significant evolution in how medical devices and in vitro diagnostic devices are regulated across the European Union. These regulations establish harmonized requirements for product registration, quality management, post-market surveillance, and technical documentation that manufacturers must navigate when placing devices on the European market.
The European Commission publishes a Rolling Plan that outlines the implementation timeline and status of the Medical Device Regulation (MDR) and In Vi…
The original regulatory framework that governed medical devices in the European Union—Directive 93/42/EEC for medical devices and Directive 98/79/EC for in vitro diagnostic devices—has been comprehensively replaced by the MDR (Regulation (EU) 2017/745) and IVDR (Regulation (EU) 2017/746). Understanding the transition timeline and implementation milestones is essential for all stakeholders involved in bringing medical devices to market in Europe.
Historical Context and Timeline Adjustments
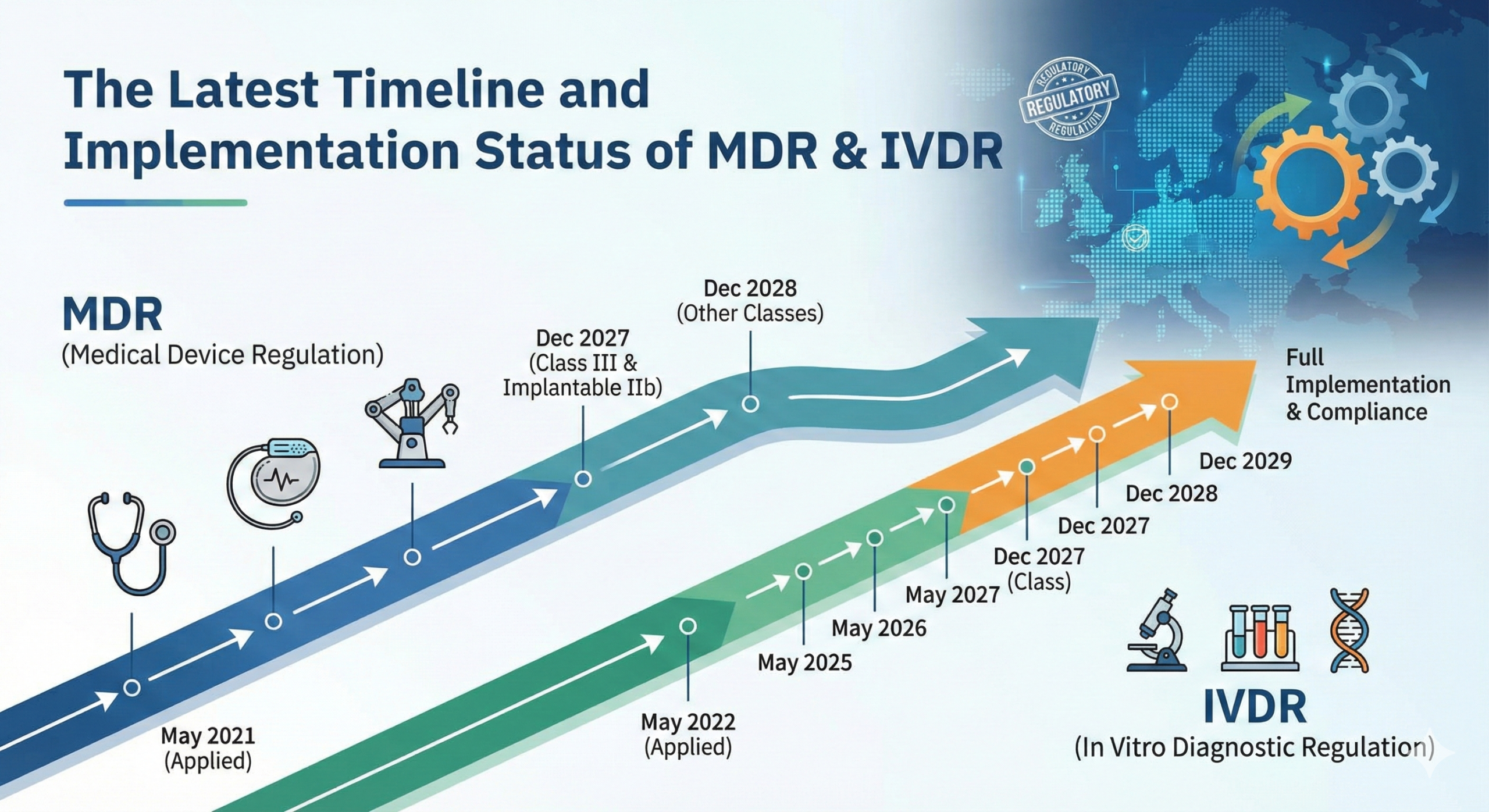
The MDR was originally scheduled to come into force on May 26, 2020. However, the COVID-19 pandemic created unprecedented challenges to the medical device industry, affecting manufacturing capacity, regulatory body operations, and the ability of manufacturers to conduct necessary compliance activities. In recognition of these challenges, the European Commission granted a one-year transition period extension. As a result, the MDR came into force on May 26, 2021, replacing the legacy Medical Devices Directive.
Similarly, the IVDR timeline underwent adjustments. The regulation was initially set to apply from May 26, 2022, but practical implementation challenges led to an additional three-year transition period, extending to May 26, 2025. This extended timeline has allowed manufacturers and notified bodies to prepare for full compliance with the new regulatory requirements.
The European Union Medical Device Database (EUDAMED), a critical digital infrastructure component for MDR and IVDR implementation, experienced more significant schedule adjustments than the regulations themselves. Originally planned for implementation at the time of MDR entry into force, EUDAMED’s rollout was substantially delayed due to technical complexity and the need for comprehensive IT system development.
EUDAMED Implementation and Phased Rollout Strategy
EUDAMED serves as the central repository for key information about medical devices and in vitro diagnostic devices within the European Union. The system manages critical data including device registrations, notified body designations, quality management system certifications, clinical evaluation reports, safety and performance summaries, and post-market surveillance information.
Rather than launching as a complete system, EUDAMED was implemented in a modular, phased approach:
| Module | Original Target | Actual Implementation | Status |
| Actor Registration Module | May 2020 | May 2023 | Operational |
| Notified Bodies Module | 2021 | June 2023 | Operational |
| Device Registration Module (Medical Devices) | 2021 | January 2024 | Operational |
| Device Registration Module (IVD Devices) | 2022 | March 2024 | Operational |
| Quality Management System Module | 2022 | September 2024 | Operational |
| Clinical Evidence Module | 2023 | Ongoing implementation | Partial functionality |
| Adverse Events and Trend Reporting | 2023 | Ongoing | In development |
This phased implementation approach enabled each module to become functional and available for user interaction as development was completed, rather than delaying the entire system until all components were ready.
Implementing Acts and Supporting Measures
The rolling plan encompasses not only the EUDAMED system but also numerous Implementing Acts and supporting measures required to operationalize the MDR and IVDR. These regulatory instruments provide detailed technical specifications, procedural guidance, and compatibility standards necessary for consistent implementation across all EU Member States.
Key Implementing Acts and measures address several critical areas. These include specifications for the reprocessing of single-use devices, which establishes the conditions and documentation requirements for manufacturers and healthcare establishments undertaking reprocessing activities. Common specifications for devices not intended for medical purposes clarify the distinction between medical devices falling under the MDR and non-medical products, a distinction that has legal and commercial implications for many manufacturers.
The designation of expert laboratories and European Union Reference Laboratories represents another crucial component of the implementation framework. These facilities provide reference standards, testing capabilities, and technical support to help manufacturers, notified bodies, and competent authorities ensure compliance with technical requirements. Under the IVDR, the designation of EU Reference Laboratories involves establishing centers of excellence capable of validating in vitro diagnostic methods and providing authoritative interpretations of performance data requirements.
Additional Implementing Acts address medical device classification rules, notified body assessment procedures, technical documentation harmonization, and IT system specifications for EUDAMED interoperability.
Transitional Arrangements and Extended Compliance Periods
To facilitate the transition from the legacy regulatory framework to the new MDR and IVDR requirements, the European Commission established transitional provisions allowing certain devices to continue under the previous regulatory regime for defined periods. These transitional arrangements were particularly important for manufacturers with devices already in the post-market phase or those requiring significant documentation revisions to meet the new standards.
For medical devices, the general transition period extends until May 26, 2025, at which point all devices must fully comply with MDR requirements, and legacy Medical Devices Directive authorizations expire. However, certain device categories and specific circumstances qualify for extended transition periods. For example, implantable devices and those subject to specific clinical evaluation requirements may benefit from additional time, though manufacturers must actively demonstrate compliance plans and progress toward MDR requirements.
The IVDR transition period extends to May 26, 2025, after which all in vitro diagnostic devices on the European market must comply with the new regulation. This alignment of the final compliance deadlines for both MDR and IVDR simplifies planning for many manufacturers dealing with product families spanning both device categories.
Current Implementation Status and Future Outlook
As of early 2025, the implementation of the MDR and IVDR represents one of the most comprehensive regulatory overhauls in the history of European medical device regulation. The core regulations have been operational for several years, enabling manufacturers to gain practical experience with new requirements such as enhanced quality management system documentation, comprehensive risk management according to ISO 14971, design and development controls aligned with ISO 13485 principles, and detailed technical documentation including safety and performance summaries intended for healthcare professionals and patients.
EUDAMED has evolved from a delayed infrastructure project into a functioning system supporting regulatory operations across the European Union. While certain modules continue to receive refinements and enhancements—particularly the Clinical Evidence Module and Adverse Events Reporting capabilities—the core functionality necessary for device registration, notified body operations, and competent authority oversight is now in place.
Manufacturers entering the European market or maintaining existing market presence must maintain current awareness of regulatory updates, competent authority guidance documents, and any additional technical standards that may be referenced or incorporated into the MDR and IVDR framework. The success of medical device regulation depends on continued cooperation among manufacturers, notified bodies, competent authorities, and the European Commission to address emerging challenges such as artificial intelligence and digital health technologies, cybersecurity requirements for connected devices, and sustainability considerations in device design and lifecycle management.
Additional Resources
For authoritative information on MDR and IVDR implementation, including the current Rolling Plan, regulatory guidance, and technical standards, stakeholders should consult the European Commission’s official documentation portal at https://ec.europa.eu/growth/tools-databases/nando/ and the EUDAMED system at https://eudamed.eu.
Educational resources and technical seminars providing detailed guidance on MDR and IVDR compliance are available through various regulatory affairs associations, professional organizations, and regulatory consulting firms. These resources help manufacturers, notified bodies, and healthcare facilities understand their obligations and implement effective compliance strategies.
Related Articles
- Guidance on Article 15 of the Medical Device Regulation (MDR) and in vitro Diagnostic Device Regulation (IVDR) regarding a 'person responsible for regulatory compliance'(PRRC)
- Full Implementation of the EU Medical Device Regulation (MDR)
- FDA Medical Device Quality System Regulation (QSR) Amendment: Current Status and Industry Impact
- Differences in Change Application Requirements for Medical Device Regulations Between Japan and the United States
- Pharmaceutical Regulations for Medical Devices in Canada
- Complaint Management in Medical Device Regulation
{kind=link}