
Understanding FDA Inspections: From Implementation to Completion
Introduction
U.S. Food and Drug Administration (FDA) inspections represent a cornerstone of regulatory compliance for pharmaceutical and medical device manufacturers. These inspections serve the fundamental purpose of ensuring product quality and safety while protecting public health. When violations are identified during an inspection, the FDA may initiate a series of regulatory actions that can significantly impact a company’s operations, ranging from advisory letters to formal enforcement proceedings.
The journey from the initial FDA inspection through potential warning letter issuance to final inspection closure carries profound implications for regulated companies. Inadequate or untimely responses to inspection findings can trigger severe consequences, including product hold orders, manufacturing suspensions, import alerts, loss of stakeholder confidence, and substantial financial impact. Therefore, maintaining robust day-to-day regulatory compliance and implementing prompt, appropriate responses to inspection findings are not merely best practices—they are essential business imperatives. Companies must sustain high quality standards continuously and prioritize consumer safety as their paramount objective.
This article provides a comprehensive examination of the FDA inspection process, from the evaluation of inspection findings through the issuance of warning letters and ultimate inspection closure. We will also explore recent regulatory developments that are reshaping the inspection landscape in 2024-2025.
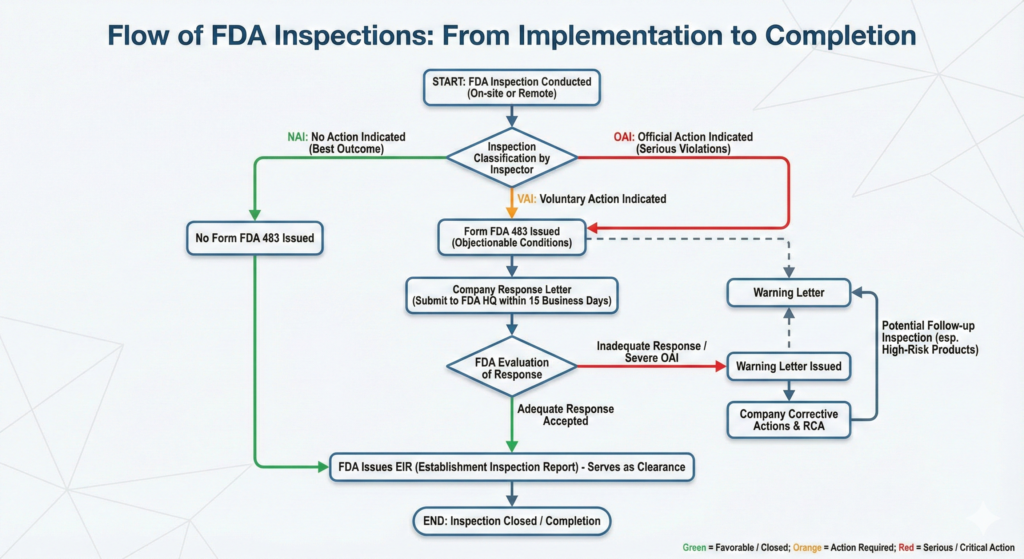
FDA Inspection Classifications
Upon completion of an FDA inspection, the agency assigns a classification that reflects the facility’s compliance status. Understanding these classifications is critical for companies to gauge the severity of findings and determine appropriate response strategies. The FDA employs three primary inspection classifications:
No Action Indicated (NAI)
NAI represents the most favorable outcome an inspected facility can receive. This classification indicates that no objectionable conditions or practices were observed during the inspection, signifying that the facility is in an acceptable state of compliance with applicable FDA regulations. In NAI situations, a Form FDA 483 (Notice of Inspectional Observations) is typically not issued to the facility management at the conclusion of the inspection.
However, it is important to recognize that receiving an NAI classification does not exempt a facility from future inspections or ongoing compliance obligations. Rather, it confirms that at the time of inspection, all observed operations, systems, and practices met FDA standards. Following an NAI inspection, the FDA will prepare an Establishment Inspection Report (EIR) documenting the inspection findings. According to current FDA policy, the final inspection classification letter is usually sent to the firm within 45 to 90 days from the close of an inspection, depending on the inspection type and complexity. If a facility does not receive this notification within the expected timeframe, it is advisable to contact the appropriate FDA district office or center to request the EIR.
Voluntary Action Indicated (VAI)
The VAI classification signifies that objectionable conditions or practices were identified during the inspection, but the agency has determined that these issues do not warrant immediate regulatory or administrative action. The FDA expects the facility to voluntarily correct the identified deficiencies without formal enforcement proceedings.
When a VAI classification is assigned, the facility typically receives a Form FDA 483 at the conclusion of the inspection, detailing specific observations made by the investigator(s). While the violations identified are significant enough to document, they do not meet the threshold of regulatory significance that would trigger Official Action Indicated (OAI) classification. Nevertheless, companies receiving VAI classifications must take these findings seriously and implement appropriate corrective and preventive actions (CAPA) to address the cited observations and prevent recurrence.
The facility is encouraged to submit a comprehensive written response to the FDA within 15 business days of the inspection’s conclusion, outlining the corrective actions taken or planned. Although a response to a Form 483 is not legally required, providing a thorough, well-documented response demonstrates the facility’s commitment to compliance and significantly reduces the risk of escalation to more serious enforcement actions.
Official Action Indicated (OAI)
OAI represents the most serious inspection classification and indicates that regulatory violations of significant consequence were identified that may warrant regulatory and/or administrative action by the FDA. An OAI classification signals that the facility is in an unacceptable state of compliance with applicable regulations. The facility may have been issued a Form FDA 483 at the conclusion of the inspection, although in some cases, particularly when violations are extremely serious, the FDA may not issue a Form 483 and may proceed directly to more formal enforcement actions.
It is crucial to understand that while all OAI-classified inspections indicate serious deficiencies, not all will result in the same enforcement outcome. The FDA may issue various types of correspondence following an OAI determination, including Warning Letters, Untitled Letters, or in cases of clinical research, Notice of Initiation of Disqualification Proceedings and Opportunity to Explain (NIDPOE) letters. The specific action taken depends on the nature, severity, and pattern of violations observed.
Understanding Form FDA 483
The Form FDA 483 serves as the primary vehicle through which FDA investigators communicate inspectional observations to facility management. This document, titled “Inspectional Observations,” lists conditions or practices observed by the investigator that, in their judgment, may constitute violations of the Federal Food, Drug, and Cosmetic Act (FD&C Act) and related regulations.
Critical Characteristics of Form FDA 483
It is important to recognize several key aspects of the Form FDA 483:
Non-Final Determination: The Form FDA 483 does not constitute a final agency determination regarding whether any condition is in violation of the FD&C Act or relevant regulations. Rather, it represents the investigator’s professional judgment based on observations made during the inspection. The FDA will consider the Form FDA 483 along with the complete Establishment Inspection Report, all documentation and evidence collected during the inspection, and the facility’s written response before making final compliance decisions.
Non-Exhaustive List: The observations noted on a Form FDA 483 are not an exhaustive listing of all objectionable conditions that may exist at the facility. FDA investigators document only what they observed during the course of the inspection. The form explicitly states: “The observations noted in this Form FDA 483 are not an exhaustive listing of objectionable conditions. Under the law, your firm is responsible for conducting internal self-audits to identify and correct any and all violations of the quality system requirements.” Therefore, companies must proactively identify and address any related or similar issues beyond those specifically cited.
Prioritization: Observations on a Form FDA 483 should be ranked in order of significance. If an observation from a prior inspection has not been adequately corrected or represents a recurring issue, this may be specifically noted on the new Form 483, potentially increasing the regulatory significance.
The Critical 15-Business-Day Response Timeline
While submitting a written response to a Form FDA 483 is not legally mandated, it is strongly encouraged and represents a critical best practice. The FDA has established a clear expectation: if the agency receives a response to the Form FDA 483 within 15 business days of the inspection’s conclusion, a detailed review of the response will be conducted prior to any decision to issue a Warning Letter or other enforcement action.
This 15-business-day timeline is calculated as follows:
- The clock begins running on the day after the Form FDA 483 is issued to facility management
- A “business day” is defined as a weekday, excluding Saturdays, Sundays, and federal holidays
- If the 15th business day falls on a weekend or federal holiday, the deadline extends to the next business day
- The response must be received by the FDA District Office by the close of business on the 15th day
Critically, companies should not wait to receive the official Establishment Inspection Report before submitting their initial response. The EIR may not arrive for weeks or even months after the inspection, and waiting for this document can result in missing the critical 15-business-day window. Failure to meet this deadline substantially increases the likelihood that the FDA will escalate the matter through more serious enforcement actions, potentially including a Warning Letter.
Components of an Effective Form FDA 483 Response
A comprehensive, well-structured response to a Form FDA 483 should address each observation individually and thoroughly. Industry best practices for Form FDA 483 responses include:
Acknowledgment and Understanding: The response should demonstrate the establishment’s acknowledgment and understanding of the FDA’s observations. Even if the facility disagrees with certain observations, the response should address them professionally and provide factual, evidence-based explanations rather than defensive or dismissive statements.
Management Commitment: The response should include clear commitment from senior leadership to address the observations. This is often accomplished through a cover letter signed by an executive officer, affirming the company’s dedication to regulatory compliance and quality.
Structured Organization: For ease of review by FDA personnel (many of whom were not present during the inspection), the response should:
- Restate each observation verbatim from the Form FDA 483
- Address each observation separately in the same sequence presented in the Form FDA 483
- Use clear headings, numbering, or table formats to link responses to specific observation numbers
Comprehensive CAPA Framework: For each observation, the response should include:
- Root Cause Analysis (RCA): A thorough investigation identifying the systemic origin of the problem—why the condition occurred, often pointing to failures in training, procedures, oversight, or system design. Superficial analyses that simply restate the observation are inadequate.
- Immediate Corrective Actions: Description of actions already taken to address the specific instances of non-compliance identified during the inspection. This should include concrete evidence such as revised procedures, completed training records, or verification testing results.
- Preventive Actions: Systemic improvements implemented to prevent recurrence of similar issues. This may include procedural enhancements, process modifications, equipment upgrades, organizational changes, or enhanced monitoring programs.
- Implementation Timeline: Realistic, specific timelines for completion of each action item, with clearly identified responsible individuals or departments. Avoid overly aggressive commitments that cannot be met; failure to deliver on promised timelines seriously undermines FDA confidence.
- Effectiveness Verification: Plans for verifying that implemented corrective and preventive actions are effective in preventing recurrence. While not explicitly required by all regulations, the FDA increasingly expects to see evidence of effectiveness checks.
Supporting Documentation: All relevant supporting evidence should be clearly referenced and included as numbered attachments. This may include updated Standard Operating Procedures (SOPs), training logs, CAPA records, validation reports, trending data, or other objective evidence demonstrating implementation of corrective actions.
Professional Tone: The response should be factual, concise, and professional throughout. Avoid defensive language, unsupported assertions, or attempts to minimize the significance of observations. Where disagreement with an observation is appropriate, it should be supported by strong factual evidence and regulatory reasoning.
Submission of the Response
The completed response package, including the cover letter, detailed responses to each observation, and all supporting documentation, must be submitted to the FDA District Office responsible for the inspection, not to the individual investigator(s) who conducted the inspection. The ultimate audience reviewing the response will typically be officials at FDA headquarters and compliance officers at the relevant FDA center (CDER, CBER, CDRH, etc.), who will assess the adequacy of the response and proposed actions.
FDA Warning Letters and Untitled Letters
When FDA observations are not adequately addressed, or when violations are of sufficient regulatory significance, the agency may issue formal correspondence in the form of a Warning Letter or Untitled Letter.
Warning Letters
Warning Letters represent formal notifications from the FDA to a company identifying specific violations of the FD&C Act or related regulations. According to the FDA’s Regulatory Procedures Manual, Warning Letters serve to achieve voluntary compliance and establish prior notice before the agency initiates more serious enforcement actions.
Key Characteristics of Warning Letters:
- Regulatory Significance: Warning Letters are issued for violations that meet a threshold of regulatory significance. They indicate that the FDA has made a determination that violations exist and that the company must take prompt corrective action.
- Prior Notice: Warning Letters provide companies an opportunity to take voluntary and prompt corrective action before the FDA initiates formal enforcement proceedings such as seizures, injunctions, or civil money penalties.
- Enforcement Warning: Unlike Untitled Letters, Warning Letters include an explicit statement warning that failure to promptly correct the violations may result in enforcement action without further notice, including product seizure, injunction, or civil penalties.
- Public Disclosure: Warning Letters are public documents. The FDA maintains a searchable database of all Warning Letters on its website, which is regularly accessed by industry stakeholders, competitors, investors, customers, and the media.
Content of Warning Letters: A Warning Letter typically includes:
- Description of specific violations observed
- Reference to applicable regulatory requirements
- A timeframe for the company to respond with corrective action plans
- A statement regarding potential enforcement actions if violations are not corrected
- Request for written response describing corrective actions taken or planned
Response Requirements: Companies that receive a Warning Letter must take immediate and comprehensive action. The letter will specify a deadline for response (typically 15 business days from receipt), and the company must implement corrective actions within the timeframes specified. The quality and thoroughness of the response are critically important. A weak, incomplete, or delayed response can result in:
- Escalation to formal enforcement actions (seizure, injunction, consent decree)
- Import alerts preventing product entry into the U.S.
- Application integrity policy issues affecting pending or future product approvals
- Referral to the Department of Justice for civil or criminal proceedings
- Significant reputational damage and loss of market confidence
Untitled Letters
Untitled Letters represent a form of regulatory correspondence used for violations that do not meet the threshold of regulatory significance required for a Warning Letter. According to the FDA’s Regulatory Procedures Manual: “An Untitled Letter cites violations that do not meet the threshold of regulatory significance for a Warning Letter.”
Distinction from Warning Letters: The key difference between Untitled Letters and Warning Letters is that Untitled Letters do not include an explicit warning statement that failure to promptly correct violations may result in enforcement action. However, this does not mean that Untitled Letters should be taken lightly. They serve as official notice that the FDA is aware of violations and expects them to be corrected.
Typical Uses: Untitled Letters are commonly used in contexts such as:
- Advertising and promotional violations that are problematic but not egregious
- First-time violations of moderate significance
- Situations where the FDA wants to document concerns without immediately escalating to Warning Letter status
Response Expectations: Although Untitled Letters do not carry the same explicit enforcement threat as Warning Letters, companies should respond comprehensively and promptly. Failure to adequately address observations in an Untitled Letter may result in escalation to a Warning Letter if violations continue or if the response is deemed inadequate.
The Establishment Inspection Report (EIR)
The Establishment Inspection Report (EIR) is the comprehensive written document prepared by the FDA investigator(s) following an inspection. The EIR serves as the official agency record of the inspection and includes:
- Detailed narrative of the inspection activities
- Description of the facility, operations, and systems observed
- Documentation of specific observations and findings
- Summary of discussions with facility personnel
- Copies or descriptions of records reviewed
- The investigator’s recommendations regarding inspection classification
Timeline for EIR Issuance: According to current FDA policy, the agency’s final inspection classification is usually communicated to the firm in a letter within 45 to 90 days from the close of an inspection. The timeline varies depending on the type and complexity of the inspection, the nature of findings, whether a response to Form FDA 483 was received, and internal FDA review processes.
In practice, some inspections may take longer than 90 days to receive final classification, particularly when:
- The inspection identified complex or serious violations requiring extensive review
- The facility is subject to OAI classification and enforcement decisions are being considered
- Multiple FDA centers or offices are involved in the review
- There are pending regulatory actions (such as Warning Letters or import alerts) that must be finalized before the EIR is released
Access to the EIR: The EIR is subject to disclosure under the Freedom of Information Act (FOIA), though certain confidential commercial information and internal FDA deliberations may be redacted. According to FDA Field Management Directive FMD-145, facilities may request a copy of their EIR, but inspections resulting in pending agency actions (such as Warning Letters, Untitled Letters, seizures, or injunctions) are not eligible for release until the inspection is formally closed and no further enforcement action is planned.
Significance of the EIR: The EIR serves multiple critical functions:
- It becomes part of the facility’s permanent FDA establishment file (e-file)
- It is reviewed in planning for future inspections of the facility
- It may be considered in evaluating pending applications that reference the facility
- It may be used in enforcement proceedings
- It provides documentation for compliance history and trending
Inspection Completion and Closure
The pathway to inspection completion depends on the classification assigned and any subsequent regulatory actions taken.
NAI Classification – Direct Completion
When an inspection receives a NAI (No Action Indicated) classification, the inspection is considered complete once the facility receives the final classification letter from the FDA. No Form FDA 483 was issued (or any observations noted were determined to be of no regulatory significance), and no further action is required from either the company or the FDA. The facility has demonstrated acceptable compliance, and routine operations may continue. The EIR will document the NAI classification and become part of the facility’s permanent FDA file.
VAI Classification – Voluntary Correction
For inspections classified as VAI (Voluntary Action Indicated), completion is achieved when:
- The facility submits a comprehensive written response to the Form FDA 483 addressing all observations
- The FDA reviews the response and determines it to be adequate
- The facility implements the corrective and preventive actions as outlined in the response
- The FDA issues a final classification letter confirming VAI status
In VAI situations, the FDA may indicate that it will verify implementation of corrective actions during a subsequent routine inspection, or in some cases, may determine that the written response and supporting documentation are sufficient without requiring a follow-up inspection.
OAI Classification with Warning Letter – Formal Closure Process
When an inspection is classified as OAI and a Warning Letter is issued, the path to closure becomes more complex and formal:
1. Written Response Required: The facility must submit a comprehensive written response to the Warning Letter within the specified timeframe (typically 15 business days), addressing each violation cited and providing detailed corrective action plans with timelines.
2. FDA Review and Evaluation: The FDA will evaluate the adequacy of the response. The agency considers whether the response:
- Demonstrates understanding of the violations
- Includes adequate root cause analysis
- Proposes corrective actions that address the violations and prevent recurrence
- Provides realistic timelines with identified responsible parties
- Shows genuine management commitment to compliance
3. Acknowledgment or Request for Additional Information: If the response is adequate, the FDA may send an acknowledgment letter indicating that the response has been reviewed and accepted. In some cases, the FDA may request additional information, clarification, or evidence of implementation.
4. Follow-Up Inspection: For serious violations or high-risk products, the FDA typically conducts a follow-up inspection to verify that corrective actions have been adequately implemented and are effective. According to FDA Field Management Directive 86, the agency establishes a goal to conduct follow-up inspections within 6 months of issuing a Warning Letter based on OAI-classified inspections, though this timeline is more consistently met for domestic facilities than for foreign facilities.
5. Close-Out Letter: Inspection closure is formally achieved when:
- The firm has responded to the Warning Letter with sufficient information demonstrating that violations have been adequately corrected, and
- A follow-up inspection (or other verified, appropriate, and reliable information) confirms that corrective actions have been adequately implemented, and
- The follow-up inspection does not reveal other significant violations
Upon meeting these conditions, the FDA may issue a “close-out letter” formally closing the Warning Letter and the associated inspection. However, not all Warning Letters receive formal close-out letters. If violations are not correctable by their nature, or if the FDA determines that ongoing monitoring is more appropriate than formal closure, a close-out letter may not be issued.
Special Considerations: Untitled Letters
When an OAI classification results in issuance of an Untitled Letter rather than a Warning Letter, the closure process is similar but generally less formal. The facility should respond comprehensively to address the cited violations and implement appropriate corrective actions. The FDA will evaluate the response and may conduct a follow-up inspection if deemed necessary. Since Untitled Letters cite violations of lower regulatory significance, the scrutiny applied to closure may be less intensive than for Warning Letters, though facilities should still treat the matter with full seriousness.
Recent Developments in FDA Inspection Practices (2024-2025)
The FDA inspection landscape is continuously evolving to address emerging challenges and leverage technological advances. Several significant developments have occurred in 2024-2025 that regulated companies should understand:
Remote Regulatory Assessments (RRAs)
On June 26, 2025, the FDA finalized its guidance document “Conducting Remote Regulatory Assessments: Questions and Answers.” This guidance formalizes the agency’s use of RRAs, which emerged as a necessity during the COVID-19 pandemic and have now become a permanent component of FDA’s regulatory oversight toolkit.
Key Aspects of RRAs:
- Definition: RRAs are remote examinations of an FDA-regulated establishment or its records to evaluate compliance with FDA requirements. Importantly, RRAs are not considered “inspections” under Sections 704(a)(1) or 704(a)(5) of the FD&C Act, which require physical entry to a facility.
- Mandatory vs. Voluntary: RRAs may be either mandatory (conducted pursuant to statutory authority such as Section 704(a)(4) of the FD&C Act) or voluntary (conducted with the establishment’s consent).
- Process: RRAs typically involve document review, virtual facility tours via livestream, and remote interviews with personnel. The FDA will notify establishments in advance whether an RRA is mandatory or voluntary and under what legal authority it is being conducted.
- Documentation: Following an RRA, the FDA may conduct a closeout meeting to present and discuss RRA observations (analogous to Form FDA 483 observations from traditional inspections). The FDA encourages establishments to submit written responses within 15 business days. The agency will prepare a written RRA report summarizing information reviewed and observations identified, which may be subject to public disclosure under FOIA.
Implications: RRAs provide the FDA with enhanced flexibility to conduct oversight activities without the logistical constraints of traditional on-site inspections. Companies should be prepared to support RRAs through:
- Ensuring document management systems allow efficient retrieval and sharing of records
- Training personnel on effective participation in remote interviews
- Developing capabilities for virtual facility tours with high-quality video feeds
- Treating RRA observations with the same level of seriousness as traditional Form FDA 483 observations
Expansion of Unannounced Inspections at Foreign Facilities
Historically, foreign drug manufacturing establishments have received advance notice of FDA inspections, allowing them time to prepare and potentially address issues before inspectors arrive. In fiscal year 2023, nearly 90% of FDA’s foreign inspections were pre-announced, in contrast with the typically unannounced inspections conducted at U.S. facilities.
On May 6, 2025, following Executive Order 14293 signed by President Trump on May 5, 2025, the FDA announced expansion of unannounced inspections at foreign manufacturing facilities producing food, essential medicines, and medical products for the U.S. market. FDA Commissioner Marty Makary stated this initiative is designed to address the “double standard” between domestic and foreign inspections and to expose non-compliant operations.
Implications: Foreign manufacturers should:
- Maintain continuous inspection readiness rather than relying on advance notice
- Ensure all records, procedures, and facilities meet FDA standards at all times
- Train personnel to respond appropriately to unannounced inspections
- Establish protocols for rapid mobilization of subject matter experts and translators
- Maintain English-language versions of critical documents
Medical Device Quality Management System Regulation (QMSR)
On January 31, 2024, the FDA issued a final rule amending the medical device Current Good Manufacturing Practice (CGMP) requirements under 21 CFR Part 820 to create the Quality Management System Regulation (QMSR). This regulation aligns U.S. requirements more closely with ISO 13485:2016, the international consensus standard for medical device quality management systems.
The QMSR takes effect on February 2, 2026, and will bring significant changes to medical device manufacturing requirements and FDA inspection approaches. The FDA is developing new inspection processes to align with QMSR requirements, which will be implemented when the rule takes effect.
Implications: Medical device manufacturers should:
- Begin transition planning immediately to ensure compliance by the February 2, 2026 effective date
- Conduct gap analyses comparing current quality systems to QMSR requirements
- Align procedures and practices with ISO 13485:2016 where applicable
- Prepare for potential changes in FDA inspection focus areas and assessment criteria
- Consider obtaining ISO 13485:2016 certification to demonstrate alignment with international standards
Quality Management Maturity (QMM) Program
The FDA’s Center for Drug Evaluation and Research (CDER) is continuing its voluntary Quality Management Maturity (QMM) Prototype Assessment Protocol Evaluation Program in 2025. This program encourages drug manufacturers to adopt advanced quality management practices that go beyond basic CGMP compliance.
From 2020 to 2024, CDER conducted several pilot programs assessing QMM at both domestic and foreign manufacturing facilities. The 2025 program uses a prototype protocol to assess establishments across five practice areas:
- Management commitment to quality
- Business continuity
- Technical excellence
- Quality culture
- Continuous improvement
While participation is voluntary, facilities that demonstrate mature quality management systems may benefit from:
- Reduced inspection frequency
- Greater regulatory flexibility
- Enhanced reputation and competitive advantage
- Improved internal quality culture and performance
Inspection Backlog and Resource Challenges
The COVID-19 pandemic led to suspension of most FDA inspections beginning in March 2020, with regular international visits resuming only in 2022. As of May 2024, approximately 2,000 pharmaceutical manufacturing firms had not been inspected by FDA since before the pandemic, including over 340 plants in India and China—major sources of drug ingredients for the U.S. market.
The FDA faces significant staffing challenges, with 225 vacancies in its inspection workforce as of June 2024—nearly four times the pre-COVID number. Factors contributing to this shortfall include extensive travel requirements, lower salaries compared to industry positions, and competition from the private sector.
Implications: Companies should be aware that:
- Inspection schedules may be unpredictable as the FDA works to address the backlog
- Facilities that have not been inspected for extended periods may receive heightened scrutiny
- The FDA is prioritizing inspections based on risk, with facilities uninspected for five or more years considered significant risk
- Companies should maintain continuous compliance readiness regardless of time elapsed since last inspection
Best Practices for Inspection Readiness and Response
Based on current FDA expectations and industry experience, companies should implement the following best practices:
Continuous Compliance Readiness
Maintain “Inspection-Ready” Status Daily: Companies should operate as though an FDA inspection could occur at any time. This means:
- Ensuring all documentation is current, accurate, and readily accessible
- Conducting regular internal audits using FDA inspection criteria
- Maintaining facilities and equipment in compliant condition
- Ensuring personnel are trained and knowledgeable regarding their responsibilities
Establish Cross-Functional Inspection Response Teams: Develop and maintain teams that include representatives from quality, manufacturing, regulatory affairs, facilities, IT, and senior management. Conduct regular drills and training exercises to ensure team readiness.
Conduct Regular Mock Inspections: Engage qualified third-party auditors or consultants with FDA inspection experience to conduct realistic mock inspections. Use findings from mock inspections to drive continuous improvement.
During the Inspection
Professional Cooperation: Provide professional, courteous cooperation with FDA investigators while maintaining appropriate boundaries. Designate experienced personnel to serve as escorts and subject matter experts.
Timely Record Production: Respond to investigator requests for records and information promptly and professionally. Document all requests and responses.
Daily Debrief Meetings: Conduct daily internal debriefs to discuss investigator observations, requests, and facility concerns. This allows real-time issue identification and response planning.
Exit Interview Preparation: Prepare carefully for the exit interview. Listen carefully to investigator observations without being defensive. Take detailed notes but avoid signing affidavits or making commitments without careful consideration.
Post-Inspection Response
Immediate Response Team Activation: Within 24-48 hours of receiving a Form FDA 483, activate investigation teams to begin gathering evidence, conducting root cause analyses, and developing corrective action plans.
Comprehensive Investigation: Conduct thorough investigations that identify true root causes, not just proximate causes. Use structured problem-solving methodologies such as fishbone diagrams, “5 Whys,” or Kepner-Tregoe analysis.
CAPA Development and Implementation: Develop robust corrective and preventive action plans that address both the specific findings and systemic issues. Assign clear ownership and realistic timelines. Implement effectiveness checks to verify that CAPAs are working.
Professional Written Response: Prepare a comprehensive, well-organized written response that addresses each observation individually. Ensure executive review and sign-off before submission.
Adherence to Commitments: Rigorously adhere to all timelines and commitments made in the response. Implement systems to track and verify completion of committed actions.
Proactive Communication: If issues arise that may delay committed actions, communicate proactively with the FDA rather than allowing deadlines to pass silently.
Conclusion
FDA inspections and the associated regulatory processes represent critical touchpoints in the life of pharmaceutical and medical device companies. Understanding the inspection classification system (NAI, VAI, OAI), the significance of Form FDA 483 observations, the implications of Warning Letters versus Untitled Letters, and the pathways to inspection closure is essential for effective regulatory compliance management.
The 15-business-day response window following receipt of a Form FDA 483 represents a critical opportunity to demonstrate commitment to compliance and prevent escalation to more serious enforcement actions. Companies that invest in thorough preparation, maintain continuous compliance readiness, and respond professionally and comprehensively to inspection findings position themselves for regulatory success.
Recent developments in 2024-2025, including the formalization of Remote Regulatory Assessments, expansion of unannounced foreign inspections, implementation of the Medical Device QMSR, and continuation of the Quality Management Maturity program, signal the FDA’s evolving approach to oversight. These changes emphasize the importance of maintaining mature, robust quality management systems that go beyond mere compliance with minimum standards.
Ultimately, companies that embrace a culture of quality, maintain transparent and professional relationships with regulators, and view inspections as opportunities for continuous improvement rather than adversarial encounters will best navigate the complex landscape of FDA oversight and sustain long-term success in bringing safe, effective products to patients.
Inspection Classification Comparison Table
| Classification | Meaning | Form 483 Issued? | Timeline for Classification Letter | Follow-Up Actions Required | Enforcement Risk |
|---|---|---|---|---|---|
| NAI (No Action Indicated) | No objectionable conditions found; facility in acceptable compliance | Usually No | 45-90 days from inspection close | None; inspection complete upon receipt of classification letter | None |
| VAI (Voluntary Action Indicated) | Objectionable conditions found but do not warrant immediate regulatory action; voluntary correction expected | Usually Yes | 45-90 days from inspection close | Written response encouraged within 15 business days; implementation of voluntary corrective actions; FDA may verify at next routine inspection | Low (if properly addressed) |
| OAI (Official Action Indicated) | Significant violations found warranting regulatory/administrative action; facility in unacceptable state of compliance | May or May Not be Issued (depending on severity) | 45-90 days from inspection close | Written response required within 15 business days; may result in Warning Letter or Untitled Letter; follow-up inspection likely within 6 months for domestic facilities; close-out letter issued upon verified correction | High; may lead to Warning Letter, import alert, application holds, or enforcement action |
Form FDA 483 Response Timeline
| Day | Action |
|---|---|
| Day 0 | Form FDA 483 issued at conclusion of inspection |
| Day 1 | Response timeline begins (day after Form 483 issuance) |
| Within 24-48 hours | Activate investigation teams; begin evidence gathering and root cause analysis |
| Days 1-10 | Conduct thorough investigations for each observation; develop corrective and preventive action plans; gather supporting evidence |
| Days 11-13 | Draft comprehensive written response; conduct internal review and obtain executive approval |
| Day 14 | Final quality check and submission preparation |
| Day 15 | DEADLINE: Written response must be received by FDA District Office by close of business |
| Ongoing | Implement committed corrective actions per stated timelines; prepare for potential follow-up inspection |
Note: Days exclude weekends and federal holidays

{kind=link}
Comment