Changes in FDA Data Integrity Observations
Background and Statistical Data
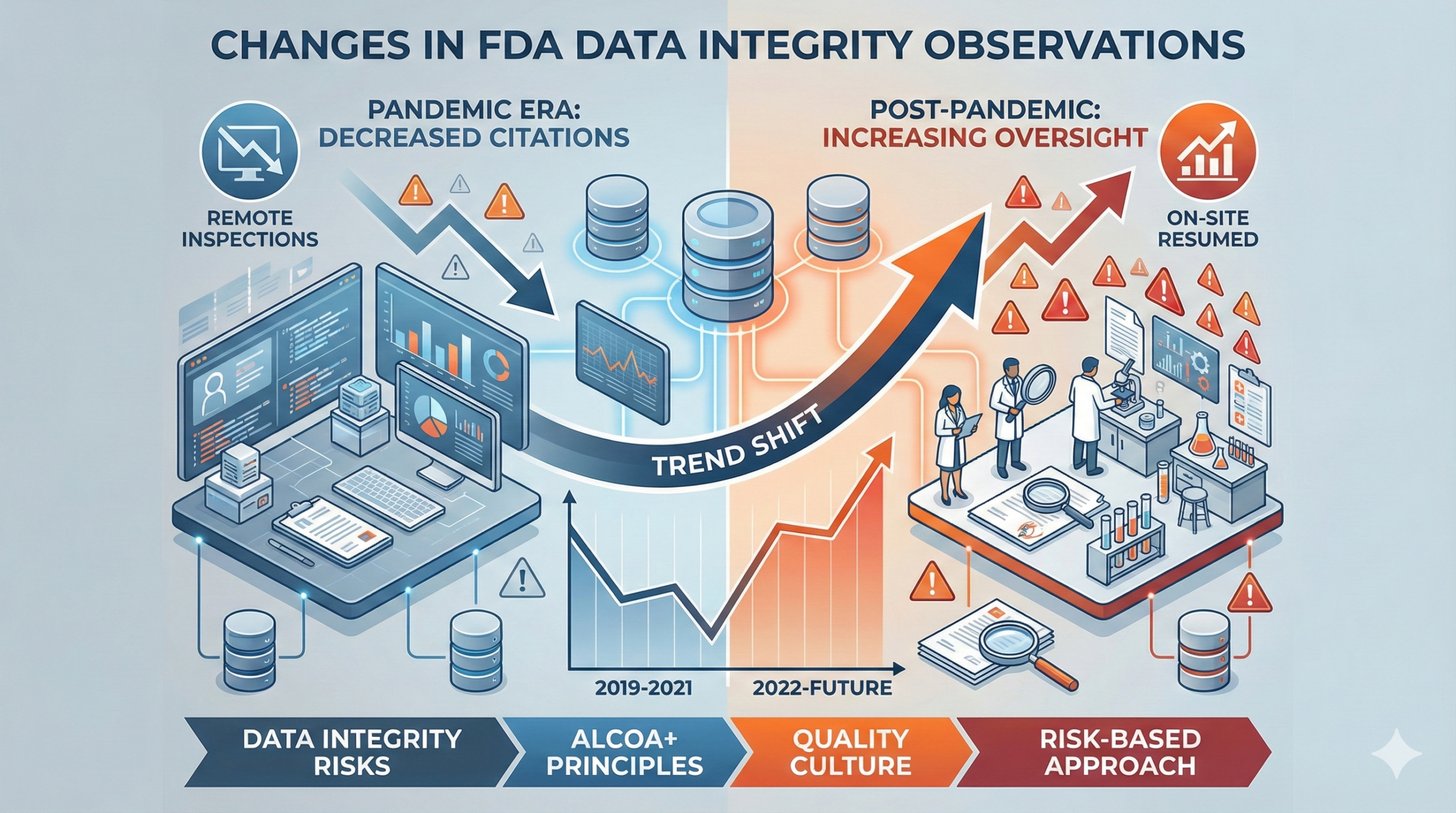
According to an article published by the Regulatory Affairs Professionals Society (RAPS), there has been a notable shift in data integrity-related observations by the FDA following changes in inspection methodologies. This trend was reported based on a presentation by Maan Abduldayem, an FDA consumer safety officer, at the 46th International Good Manufacturing Practice (GMP) Conference.
The number of data integrity observations has shown the following trajectory: In fiscal year (FY) 2019, data integrity issues were cited in 32 Warning Letters (W/L). This decreased to 24 W/L in FY2020, and further declined to merely 5 W/L in FY2021. This dramatic reduction is primarily attributed to changes in inspection approaches, particularly the shift to remote inspections necessitated by the COVID-19 pandemic.
Remote inspections inherently limit critical activities essential for evaluating data integrity, such as direct on-site observations, detailed document review, spontaneous employee interviews, and unannounced record inspections. Consequently, rather than indicating an actual decrease in violations, this trend more likely reflects a temporary reduction in detection capability. Indeed, as the pandemic has subsided, the FDA has progressively resumed on-site inspections, and observation frequencies are expected to increase accordingly.
Relationship Between Data Integrity and Patient Safety and Product Quality
When data integrity is not assured, direct impacts on patient safety and product quality assurance ensue. Understanding this relationship is essential for recognizing the critical importance of data integrity in pharmaceutical manufacturing.
In quality control (QC) laboratories conducting quality testing, data falsification or inaccurate recording—whether intentional or inadvertent—creates risks of substandard products reaching the market that should not have been released. A crucial point here is that while intentional falsification is undoubtedly most serious, lapses in data reliability due to inadvertent errors (human errors) are equally problematic. From the perspective of patient safety impact, whether data reliability is compromised intentionally or inadvertently makes no difference in the resulting risks.
Statistically, inadvertent errors account for approximately 80% of all data integrity failures. Specific examples include transcription errors from handwritten records, calculation mistakes, equipment operation errors, and measurement recording errors. Furthermore, intentional acts do not necessarily stem from malicious intent. For instance, operators may “correct” raw data believing it to be right, or delete outliers judging them as “obvious instrument errors.” These situations arise from misconceptions, assumptions, memory lapses, inadequate understanding of Standard Operating Procedures (SOPs), or ambiguous and unclear SOPs themselves.
From a product quality assurance perspective, the integrity of manufacturing records, batch records, validation records, and stability testing data is indispensable. When these records contain deficiencies or falsifications, it becomes impossible to verify that manufacturing processes were properly controlled, thereby precluding quality assurance of the product.
FDA’s Regulatory Response
Situations where data integrity cannot be confirmed have profound implications for FDA regulatory decisions. When the FDA cannot verify patient safety and product quality, it cannot approve product release. This requirement is based on the Federal Food, Drug, and Cosmetic Act (FD&C Act) and Current Good Manufacturing Practice (CGMP) regulations (21 CFR Parts 210, 211).
In Pre-Approval Inspections (PAI), data integrity issues have particularly severe consequences. PAIs are conducted as part of the review process for New Drug Applications (NDA) or Abbreviated New Drug Applications (ANDA) to verify that manufacturing facilities described in application submissions meet appropriate GMP standards. When significant data integrity deficiencies are discovered during such inspections, the FDA must question the reliability of the data in the application submission itself. Consequently, the agency cannot determine whether to accept the application data, affecting approval or non-approval determinations within the timeframes established by the Prescription Drug User Fee Act (PDUFA). In worst-case scenarios, a Complete Response Letter (CRL) may be issued, resulting in approval delays or rejections.
International Regulatory Harmonization and Data Integrity
The importance of data integrity is widely recognized internationally. The Pharmaceutical Inspection Co-operation Scheme (PIC/S) issued “Good Practices for Data Management and Integrity in Regulated GMP/GDP Environments” (PI 041-1) in 2021, providing international guidance on data integrity. This guidance is founded on ALCOA+ principles: Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, and Available.
The European Medicines Agency (EMA) and the Medicines and Healthcare products Regulatory Agency (MHRA) have issued similar guidance, and the World Health Organization (WHO) also provides data integrity guidance. In Japan, the Pharmaceuticals and Medical Devices Agency (PMDA) and the Ministry of Health, Labour and Welfare issued “Guidance on Basic Concepts Regarding Data Integrity for Pharmaceuticals” in 2019, aligning with international standards.
Practical Approaches to Ensuring Data Integrity
Ensuring data integrity requires both technical and organizational measures. Technical measures include appropriate implementation of electronic record and electronic signature systems (compliant with 21 CFR Part 11 and EU Annex 11), establishment of audit trails, proper access control settings, and implementation of data backup and disaster recovery plans.
Organizational measures include comprehensive employee education and training programs on data integrity, creation and periodic review of clear and executable SOPs, cultivation of quality culture, regular internal audits and self-inspections, and establishment of appropriate Corrective and Preventive Action (CAPA) systems for data integrity violations.
Particularly for preventing inadvertent errors, consideration of human factors is essential. Systematic approaches are required, including work environment improvements, workload optimization, implementation of double-check systems, and construction of error-proofing (Poka-Yoke) mechanisms.
Future Outlook
Following the pandemic, the FDA has steadily resumed on-site inspections while adopting a risk-based approach. Since 2023, FDA inspection activities have been approaching pre-pandemic levels, and data integrity observations are reportedly trending upward again. Companies are encouraged to proactively identify and correct issues that may have been overlooked during the remote inspection period.
Furthermore, with the advancement of digital transformation (DX), new technologies are being introduced, including data integrity monitoring systems utilizing artificial intelligence (AI) and machine learning (ML), blockchain technology for preventing data falsification, and cloud-based quality management systems. However, the introduction of these new technologies must meet Computer System Validation (CSV) requirements.
Ensuring data integrity is not merely a matter of regulatory compliance, but a fundamental responsibility for protecting patient safety and assuring product quality. Pharmaceutical companies are required to establish and maintain robust data integrity systems through continuous improvement activities.
Document Information:
- Original Language: Japanese
- Translation Date: January 2026
- Topic: FDA Data Integrity Regulatory Trends
- Target Audience: Pharmaceutical industry professionals, quality assurance specialists, regulatory affairs professionals
{kind=link}
Comment