Implementation of IOQ (Installation and Operational Qualification)
Growing Trend of Combined IQ and OQ Implementation
In recent years, there has been an increasing trend toward conducting Installation Qualification (IQ) and Operational Qualification (OQ) simultaneously. This practice became formally acceptable following the revision of the PIC/S GMP Annex 15 “Qualification and Validation” in 2015, which came into effect on October 1, 2015.
According to Section 3.10 of the revised Annex 15, Operational Qualification normally follows Installation Qualification. However, depending on the complexity of the equipment, it may be performed as a combined Installation/Operation Qualification (IOQ). Furthermore, the documentation for IQ and OQ may be consolidated into a single document. This flexibility represents a significant evolution in validation practices, allowing pharmaceutical manufacturers to streamline their qualification processes while maintaining compliance with Good Manufacturing Practice (GMP) requirements.
Installation Qualification (IQ) Requirements and Scope
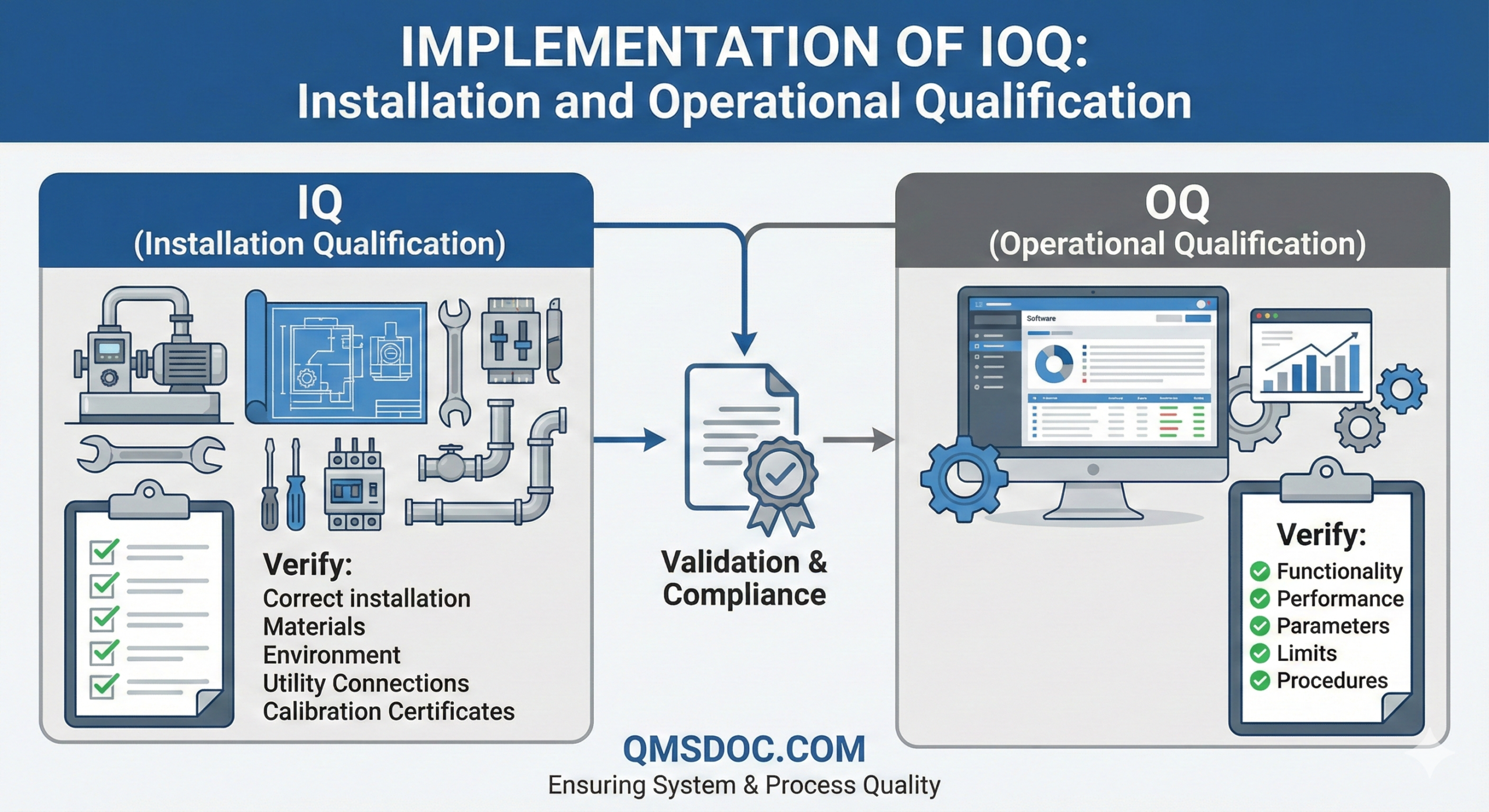
Installation Qualification should be performed on equipment, facilities, utilities, or systems within pharmaceutical manufacturing operations. The fundamental purpose of IQ is to provide documented verification that the system has been delivered, installed, and configured according to predetermined specifications and manufacturer’s requirements.
IQ activities typically include, but are not limited to, the following elements:
Cross-verification of components, instruments, equipment, piping, and services against engineering drawings and detailed specifications to confirm correct installation. This verification ensures that all physical aspects of the system match the approved design documentation and that no deviations exist between the planned configuration and the actual installed state.
Verification that installation has been completed correctly according to predefined conditions and specifications. This includes confirming that all utilities are properly connected, that spatial requirements are met, and that the installation complies with relevant safety standards and local regulations.
Collection and verification of operational procedures, work instructions, and maintenance requirements from suppliers. This documentation forms the foundation for ongoing operations and ensures that the pharmaceutical manufacturer has complete information necessary for proper system management throughout its lifecycle.
Calibration of measuring instruments and control devices. All critical measurement and control equipment must be calibrated using traceable standards to ensure accuracy and reliability of the system’s performance monitoring capabilities.
Verification of construction materials used in the system. This is particularly important for pharmaceutical applications where product contact surfaces must be verified to be appropriate materials (such as stainless steel grades 316L or 304 for most applications) that will not adversely affect product quality, contaminate the product, or compromise system integrity.
Operational Qualification (OQ) Requirements and Scope
Operational Qualification normally follows the successful completion of Installation Qualification. However, as mentioned above, depending on the complexity of the equipment, it may be performed simultaneously as IOQ. The decision to combine or separate these qualification stages should be based on a risk assessment that considers factors such as equipment complexity, automation level, criticality to product quality, and regulatory expectations.
OQ activities typically include the following elements:
Tests developed based on knowledge of the process, system, and equipment to ensure the system operates as designed. These tests should challenge the system’s functionality across its intended operating range and verify that all critical functions perform according to specifications. The test protocols should be derived from the Functional Specification (FS) and User Requirements Specification (URS) to ensure complete coverage of all critical requirements.
Testing to confirm operational upper and lower limits, including “worst case” conditions. This testing is essential to establish the validated operating range and to demonstrate that the system can perform reliably even under challenging or extreme conditions that may be encountered during routine operations. Worst-case testing might include maximum and minimum processing volumes, extreme environmental conditions within acceptable ranges, or maximum throughput scenarios.
Upon successful completion of OQ, it becomes possible to finalize Standard Operating Procedures (SOPs), cleaning procedures, operator training programs, and preventive maintenance requirements. This represents a critical milestone in the equipment lifecycle, as it signifies that the system is ready for routine operation and that all necessary operational infrastructure can be completed and implemented.
Omission of DQ and PQ in Modern Practice
In contemporary pharmaceutical manufacturing, it has become common practice to omit certain qualification stages for structural equipment and facilities. Design Qualification (DQ) is now rarely performed because most structural equipment consists of catalog products or off-the-shelf items rather than custom-designed systems. When equipment is procured from established manufacturers with proven designs, the need for extensive design qualification is significantly reduced.
According to PIC/S GMP Annex 15, Performance Qualification (PQ) may be performed in conjunction with OQ or Process Validation. This flexibility allows pharmaceutical manufacturers to optimize their validation strategies based on risk assessment and the specific characteristics of their systems. Consequently, for structural equipment validation, DQ and PQ are increasingly omitted or integrated with other validation activities, streamlining the overall qualification process while maintaining appropriate quality assurance.
Performance Qualification (PQ) When Conducted
When Performance Qualification is performed as a separate stage, it should be conducted after the completion of IQ and OQ. However, as noted in Annex 15, in some cases it may be appropriate to perform PQ in conjunction with OQ or Process Validation, particularly for well-understood systems or when extensive development data supports the approach.
PQ activities typically include the following elements:
Testing using actual production materials, qualified substitute materials, or simulant products at worst-case batch sizes that demonstrate behavior equivalent to routine operating conditions. The use of actual production materials or qualified substitutes ensures that the qualification accurately reflects real-world manufacturing scenarios and provides confidence that the system will perform consistently during commercial production.
Demonstration of appropriate sampling frequency rationale used to confirm the process is in a state of control. The sampling plan should be scientifically justified based on statistical principles, risk assessment, and knowledge of process variability. Adequate sampling is essential to detect process variations and ensure that critical quality attributes remain within acceptable limits.
Testing that covers the intended operating range of the process, unless there is documented evidence from the development stage that supports the operating ranges. The testing program should encompass all parameters that may affect product quality and should demonstrate that the process consistently produces products meeting predetermined specifications and quality attributes across the full range of anticipated operating conditions.
Factory Acceptance Testing (FAT) and Site Acceptance Testing (SAT)
A crucial prerequisite before implementing DQ, IQ, OQ, and PQ is the development of User Requirements Specifications (URS) and the execution of Factory Acceptance Testing (FAT) and Site Acceptance Testing (SAT). These early-stage activities establish the foundation for successful equipment qualification and validation.
Factory Acceptance Testing (FAT)
Equipment incorporating novel or complex technologies may be evaluated by the vendor prior to shipment to the pharmaceutical manufacturer (before IOQ implementation). This factory-based testing serves multiple critical purposes in the qualification lifecycle.
Before equipment installation, vendor compliance with URS and Functional Specifications should be confirmed at the factory. This pre-shipment verification provides early detection of any discrepancies between requirements and actual equipment capabilities, allowing issues to be resolved before the equipment arrives at the pharmaceutical site.
When appropriate justification exists, and it can be demonstrated that transportation and installation will not affect functionality, document reviews and certain tests may be performed during FAT or other stages, eliminating the need to repeat IQ/OQ testing on-site at the pharmaceutical facility. This approach can significantly reduce qualification timelines and costs while maintaining appropriate quality assurance. The key principle is that if vendor FAT is properly executed with appropriate rigor and documentation, the scope of IOQ implementation can be substantially reduced.
The extent to which FAT can substitute for on-site qualification depends on several factors, including the complexity of the equipment, the thoroughness of FAT protocols, the similarity between factory and site conditions, and regulatory expectations. A risk-based approach should guide decisions about which tests must be repeated on-site and which FAT results can be leveraged to streamline the qualification process.
Site Acceptance Testing (SAT)
FAT can be complemented by implementing SAT after equipment receipt at the manufacturing site. SAT provides confirmation that the equipment was not damaged during transportation and that it functions properly when connected to site utilities and integrated into the actual manufacturing environment. SAT typically covers verification of utility connections (electrical power, compressed air, water systems, etc.), confirmation of proper equipment function under site conditions, and documentation that the equipment is ready for formal qualification activities.
Evolution of Equipment Qualification Responsibility
The author frequently assists pharmaceutical companies and venture enterprises in implementing IOQ. Common questions concern the differences between FAT/SAT and IOQ, as well as the distinction between PQ and Process Validation. These questions reflect the evolving understanding of qualification responsibilities and practices in the pharmaceutical industry.
Historically, it was considered mandatory for qualification activities to be performed by the pharmaceutical company itself. In many cases, pharmaceutical companies requested vendors to create qualification documentation, but this documentation was formatted to appear as if the pharmaceutical company had performed the work. This practice reflected regulatory expectations and industry norms that emphasized pharmaceutical company ownership of qualification activities.
However, modern regulatory thinking recognizes that the identity of who performs the qualification activities is less important than ensuring that the quality of the products manufactured using the equipment is assured. The critical question is not “who performed the qualification?” but rather “does the qualification provide appropriate assurance that the equipment will consistently operate as intended and produce quality products?”
In most contemporary cases, pharmaceutical companies delegate IOQ implementation to equipment vendors. This approach makes practical sense because vendors possess intimate knowledge of their equipment, have greater technical expertise regarding equipment functionality and performance characteristics, and can often perform qualification activities more efficiently and effectively than pharmaceutical companies. The pharmaceutical company’s role evolves to one of oversight, review, and approval rather than direct execution.
Current Industry Practice and Quality Assurance Perspective
The fundamental principle in modern pharmaceutical equipment qualification is that the ultimate responsibility for product quality remains with the pharmaceutical manufacturer, regardless of who executes the qualification activities. Whether qualification is performed by the vendor, a third-party consultant, or the pharmaceutical company’s own personnel, the manufacturer must ensure that the qualification is thorough, scientifically sound, and provides appropriate assurance that the equipment will consistently perform as required.
This evolution in thinking aligns with modern quality risk management principles and Quality by Design approaches promoted by ICH Q8, Q9, Q10, and Q11 guidelines. The focus has shifted from rigid procedural requirements to flexible, science-based approaches that achieve the fundamental objective of ensuring equipment reliability and product quality while optimizing resource utilization and project timelines.
In practice, most pharmaceutical companies now request vendors to perform IOQ activities, recognizing that vendors are often best positioned to conduct these activities effectively. The pharmaceutical company maintains quality oversight through protocol review and approval, witness of critical testing activities, review and approval of qualification reports, and integration of qualification results into the overall validation program and quality management system.
This collaborative approach between pharmaceutical manufacturers and equipment vendors, with clear definition of roles and responsibilities, represents best practice in modern pharmaceutical equipment qualification. It optimizes the use of technical expertise, streamlines qualification processes, reduces costs and timelines, and ultimately provides robust assurance that equipment will consistently produce quality pharmaceutical products.
Regulatory References:
- PIC/S GMP Annex 15: Qualification and Validation (Revised 2015, effective October 1, 2015)
- EU GMP Annex 15: Qualification and Validation (EudraLex Volume 4)
- ICH Q8: Pharmaceutical Development
- ICH Q9: Quality Risk Management
- ICH Q10: Pharmaceutical Quality System
- ICH Q11: Development and Manufacture of Drug Substances
Note: This article represents general guidance on qualification practices. Specific requirements may vary based on product type, manufacturing complexity, regulatory jurisdiction, and company-specific quality management systems. Organizations should consult relevant regulations and guidance documents applicable to their specific circumstances and maintain compliance with all applicable regulatory requirements.
{kind=link}
Comment