| 規 格/標 題 | |
|---|---|
| IEC62304:2006 Medical device software - software life cycle process | 医療機器ソフトウェア-ソフトウェアライフサイクルプロセス |
| IEC80001-1:2010 Application of risk management for IT-networks incorporating medical devices- Part1: Roles, responsibilities and activities | 医療機器に結合するITネットワークへのリスクマネジメントの適用 |
| IEC/TR 80002-1:2009 Medical devices software-Part:1 Guidance on the application of ISO 14971 to medical device software | 医療機器ソフトウェア-第一部:医療機器ソフトウェアへのISO14971の適用の手引き |
| IEC60601-1-4:2000 Medical electrical equipment – Part 1-4 General requirements for safety-Collateral standard Programmable electrical medical system | 医用電気機器 – 第1-4部: 安全性の一般要求事項 – 副通則:プログラマブル電気医用システム |
IEC-62304とは
IEC62304(Medical device software – software Life Cycle Processes)は、 医療機器ソフトウェアの安全性の向上を目的として、ソフトウェア開発及び保守に関する要求項目を規定した規格である。
2006年5月に発行された医療機器ソフトウェアのライフサイクルプロセスの枠組みを示した最初の国際規格である。
IEC 62304は「医療機器ソフトウェア」の開発と保守に関するプロセスを規定している。
2015年6月にAmendment 1 が加わった IEC 62304の改訂版Edition 1.1が発表された。
改訂版にはレガシーソフトウェアの扱い、一般的なソフトウェア欠陥の特定と回避等が新しく加わった。
Ed.1.1は、閲覧だけならば日本工業標準調査会ホームページ(http://www.jisc.go.jp)で参照できる。
IEC62304は
- 安全要求を実現するための、ソフトウェア開発&保守手順標準
- 医療用ソフトウェアのみをターゲット
- ライフサイクルプロセスの規格
- ソフトウェア品質を確保するための製造者のアクティビティについて規定
- 特定の開発モデルを想定してはいない
本規格は、ソフトウェアの試験に関する要求を含んでいるが、その詳細な要求をしていない。欠陥を作らないよう適切なプロセス(&タスク)を実施しエビデンスを文書化することを重視している。その理由は、「ソフトウェアの試験だけでは、動作時の安全性を判定する上で十分ではない」との考え方に基づく。
本規格では、医療機器ソフトウェアに内在する危険度に応じて3段階の「安全クラス」を定義し、安全性クラスに応じてライフサイクルの各プロセスにおいて最低限実施すべき一連のアクティビティと各工程で実施するタスクを規定している。
製造者は、初めにリスクマネジメントに基づきソフトウェアに起因するハザードが人に及ぼす影響を安全性クラスに分類し、リスク管理対策を定義、文書化することを求められる。
次に、適切な品質マネジメント、ソフトウェアエンジニアリングの下で、開発や検証等における一連のプロセスの中でリスクに応じた作業を実施、記録、管理することを求められる。
現状、欧米を始めとする各国の規制においてIEC62304が適用されるようになってきており、医療機器ソフトウェアの安全性確保のための国際的な規格要求はIEC62304 に集約されつつある。
規制要件としてのIEC-62304
医療機器ソフトウェア開発の現場において、IEC 62304は、国際的に重要な位置づけの規格として認識されている。
IEC 62304は、2006年5月に発行され、日本では2012年にJIS化(JIS T 2304)された。
2014年11月に施行された医薬品医療機器法第12条第2項において参照される「最新のライフサイクルモデル」である 。
2017年3月1日官報にて、IEC 62304 Ed. 1.1 (IEC 62304:2006 + Amd.1:2015)に対応した“JIS T 2304:2017が公示された。
今後3年間で、この改正へ対応することが求められる。
リスク期間については、規格自体には記載はない。(IEC 62304 Amd1には記載があり、3年以内のリスクを推奨している。)
本邦において、2017年11月25日より、IEC 62304(医療機器ソフトウェア ‐ ソフトウェアライフサイクルプロセス)が実質的な規制要件となった。
- プログラム(ソフトウェア)を用いた医療機器はJIS T 2304への適合の確認をする。
- 承認または認証を行う場合は、申請書添付資料において、JIS T 2304への適合を説明する。
- 一般医療機器(クラス1)も同様に確認が必要。
- 2017年11月24日までに設計が完了している医療機器であっても、 JIS T 2304等への適合を満たすための措置を講じること。
日本以外でも欧州・北米・中国などにおいて医療機器申請時にIEC 62304に基づくソフトウェア開発の証拠が必要である。
つまりIEC 62304に従って「医療機器ソフトウェア」を開発しなければ、国内外においてソフトウェアを搭載した医療機器(単体プログラムを含む)を販売することができない。
米国FDAにおいても2008年7月にRecognized Consensus Standardと認定された。
IEC62304の三大原則
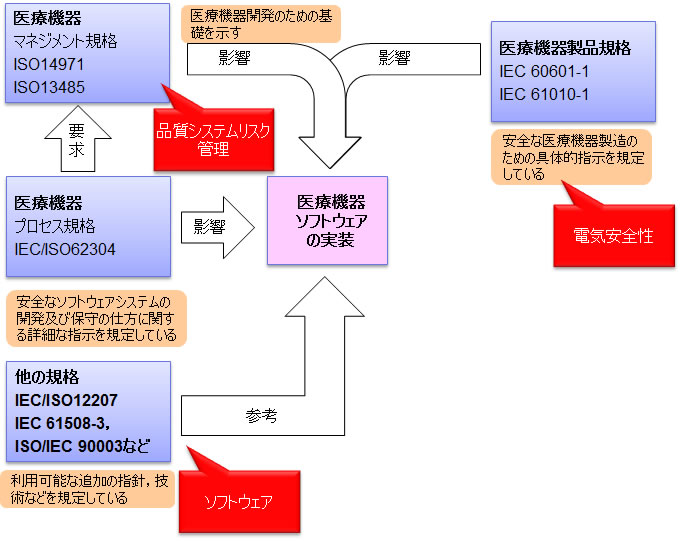
IEC62304では、医療機器ソフトウェアの安全性を向上させる三つの大原則として、「リスクマネジメント、品質マネジメント、ソフトウェアエンジニアリング」を挙げている。
- リスクマネジメント:ISO14971に適合するリスクマネジメントプロセスの適用を要求
- 品質マネジメント:ISO13485 などの適切な品質マネジメントシステムの適用を要求
- ソフトウェア・エンジニアリング:この規格(IEC 62304)への適合を要求
ソフトウェアの安全性は、ソフトウェア自体が持つ特性ではなく、ソフトウェアとその外部(ハードウェアなど)とのかかわりの中で現れるものとされている。そのため、この規格は、医療機器向けのリスクマネジメントの規格であるISO 14971の力も借りて、 ソフトウェアが関わるリスクを管理しながら、安全要求に対して品質の高いソフトウェアを提供することを目指している。
IEC-62304対応課題
医療機器企業では、品質マネジメントやIEC62304等の規格適合を意識した開発の必要性を認識しているが、これらの規格に対応できる人材が不足している点、規格書だけ読んでも対応すべき内容や方法が分からない点が大きな壁となっている。
現状では、ソフトウェアに重点を置いた開発が行われておらず、ソフトウェアの設計・開発などの各フェーズでの検証が不十分となる上、設計開発記録などの技術文書構築が行われない傾向にある。
今後どのようにソフトウェアシステム(QMS)を構築していくかが問題である。
ソフトウェアを搭載した医療機器の海外展開の問題点
- IEC62304、FDAガイダンスに対応しなければ海外展開できない。
- 日本向け製品では海外規格を意識せずに開発をしている。
- これまで日本において医療機器ソフトウェア開発に関する規制がなかったため、品質マネジメントの基準や手順は企業任せになってしまい、企業によって品質のバラツキが大きくなる。
- 自社製品を海外当局へ薬事申請を行う場合、ソフトウェア設計開発記録を初めから起こしなおす、又は再編集しないと要求事項に適合しない。
- IEC62304規格の適合確認を受けていた製品に対して、FDAから適合していないとの厳しい指摘を受けた。
IEC62304対応文書構築の問題点
- 企業では、品質マネジメントやIEC62304等の規格適合を意識した開発の必要性を認識しているが、規格書だけ読んでも対応すべき内容や方法が分からない。
- 規格書を読んでも、どこまでやるべきなのかの範囲が分からない。
- 規格の詳細の内容が不明なまま、IEC62304文書構築を行っている。
医療機器企業におけるソフトウェア開発の問題点
人員不足
- ソフトウェア開発担当者が少なく、開発期間も短いことから、まともな設計資料を作っていない。
- 開発記録の作成は、ソースプログラムの構想・構築とは別工程として時間を要するため、十分な開発人材をあてがうことができない。
- 人員が慢性的に不足しており、開発手法やISO、IEC規格に基づいた教育・トレーニングを受けさせる時間的余裕がない。
- トレーナーの育成ができない。
規程・手順書の不備
- 現状、ソフトウェアについての管理手順がない。
- ISOの書類が形式的になりがちである。そもそもISOのソフトウェアの管理手順が存在していない。
- 製品の実装、期日通りの納品を優先させるために、製品の開発手法や品質の標準化は置き去りにされている。
- 開発を安全に行うには、構成管理や変更管理をしっかり行うプロジェクトマネージメント手法を取り入れる必要があるが、個々の能力と責任感、職人かたぎに頼ったプロジェクト推進が主流の日本的開発スタイルからの切替えは難しい。
設計開発記録の不備
- ソフトウェアに重点を置いた開発があまり行われず、ソフトウェアの設計・開発などの各フェーズでの検証が不十分となる上、設計開発記録などの技術文書構築が行われない傾向にある。
- ソフトウェアの実装ありきで、設計開発記録を用いた分析、エビデンスの保管というフローが存在していない。
- 連動して開発されたソフトウェアの一連の設計開発書が存在しない。存在したとしてもISOやIEC規格の要求事項とは掛け離れた記録でしかない。
- ドキュメンテーションが実際の開発フローに準じていない。
ソフトウェア設計開発の課題
IEC-62304とキーとなる医療機器関連標準との関係
IEC62304 目次
まえがき
序文
1.適用範囲
1.1 目的
1.2 適用分野
1.3 他の規格との関係
1.4 適合
2.引用規格
3.用語及び定義
4.一般要求事項
4.1 品質マネージメントシステム
4.2 リスクマネジメント
4.3 ソフトウェアの安全性分類
4.4 レガシーソフトウェア
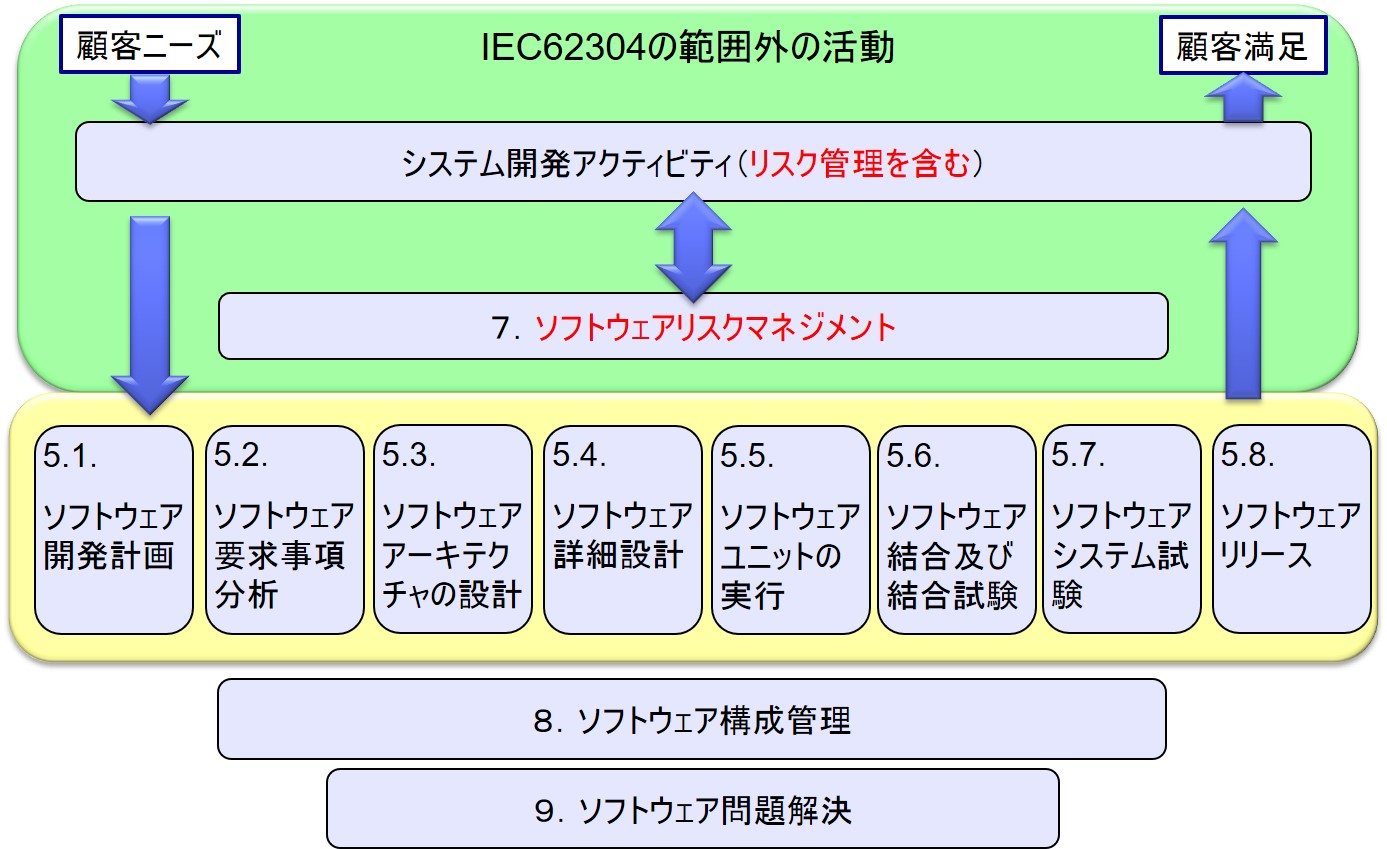
5.ソフトウェア開発プロセス
5.1 ソフトウェア開発プランニング
5.2 ソフトウェア要求事項分析
5.3 ソフトウェアアキテクチャの設計
5.4 ソフトウェア詳細設計
5.5 ソフトウェアユニットの実行
5.6 ソフトウェア結合及び結合試験
5.7 ソフトウェアシステムの試験
5.8 ソフトウェアリリース
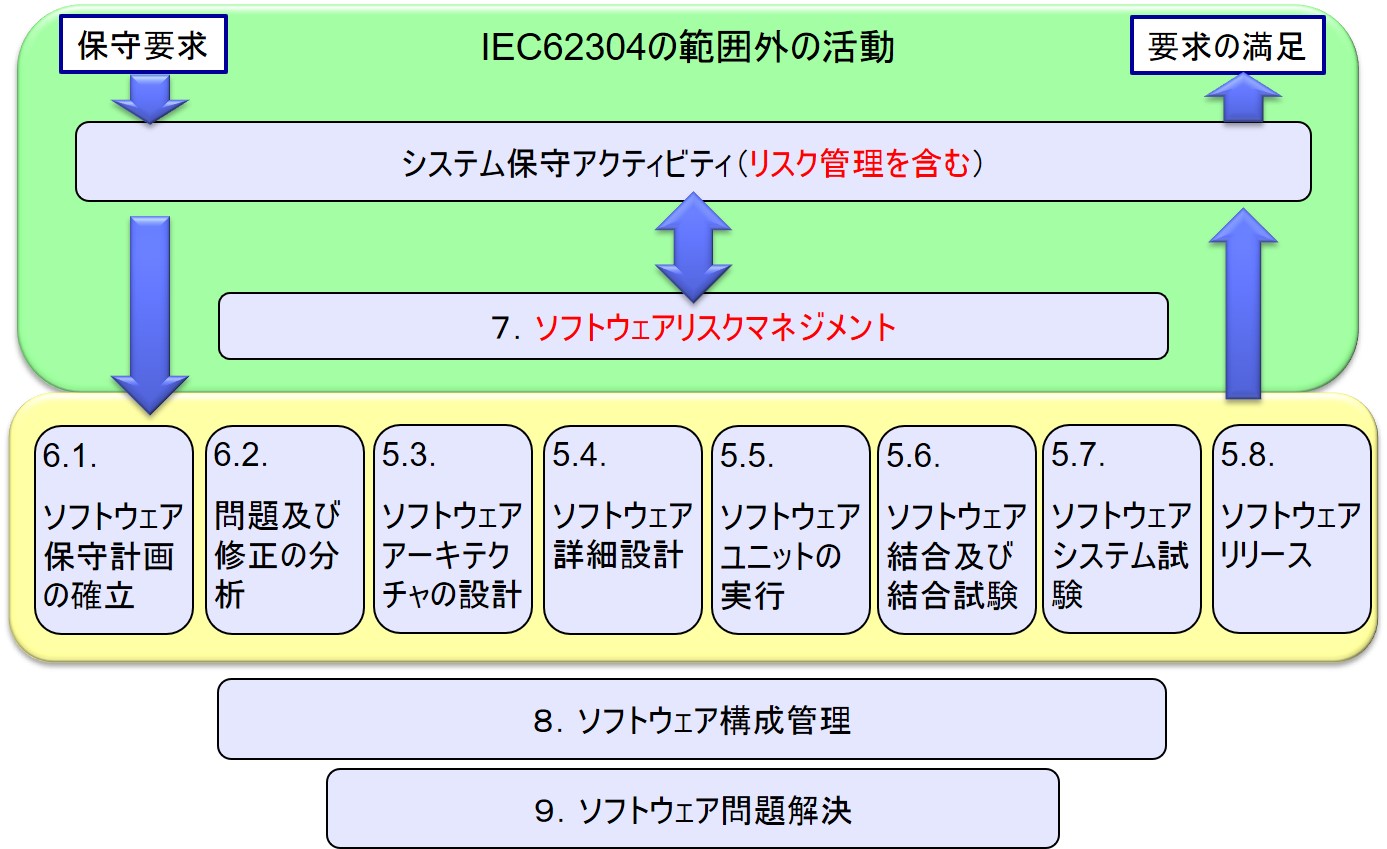
6.ソフトウェア保守プロセス
6.1 ソフトウェア保守計画の策定
6.2 問題及び修正の分析
6.3 修正の実施
7.ソフトウェアリスクマネジメントシステム
7.1 危険な状況を誘発するソフトウェアの分析
7.2 リスク管理策
7.3 リスク管理策の検証
7.4 ソフトウェア変更のリスクマネジメント
8.ソフトウェア構成管理プロセス
8.1 構成の識別
8.2 変更管理
8.3 構成状態アカウンティング
9.ソフトウェア問題解決プロセス
9.1 問題報告書の作成
9.2 問題の調査
9.3 関係当事者への通知
9.4 変更管理プロセスの使用
9.5 記録の維持
9.6 問題の傾向分析
9.7 ソフトウェア問題解決の検証
9.8 試験文書の内容
附属書A 論拠
附属書B この規格の規定の手引き
附属書C 他の規格との関係
附属書D 実施
参考文献
ソフトウェア開発プロセスの概観
ソフトウェア保守プロセスの概観