
2014年11月25日から、薬事法が一部改正される。…
これにより、現在の「薬事法」という名称から、「医薬品、医療機器等の品質、有効性および安全性の確保等に関する法律」 (医薬品医療機器等法)という名称に変更される。
この法律の施行に向けて、新法の「施行令」、「施行規則」、「新QMS省令」、製造販売業者の許可基準たる「体制省令」、新法下におけるQMS調査の単位となる「製品群区分省令」などがまとめて公布された。
さらに各種運用通知等の検討も進められている。
改正法では、医療機器の「機械器具等」の範疇に、「ソフトウェア(プログラム)」が追加されるなど医療機器に関して大きな変更がなされた。
そのため、「体制省令」に基づく組織体制の整備をはじめ、新QMS省令に準拠した手順書類の確立、また製造業の登録や、既存品目の新法移行手続き(記載整備)、そして新法下における更新QMS調査に向けた準備など、医療機器の製造販売業者、製造業者には各種の対応が求められることになった。
改正法の施行に伴い、製造販売業の許可要件であるGQP省令が抜本的に見直され、QMS省令に包括される。つまり、これまでのQMS省令は、製造業者にのみ適用されてきたが、新QMS省令は製造販売業者にも適用されることとなった。
薬事法改正(医薬品医療機器等法)
医薬品、医療機器等の安全かつ迅速な提供の確保を図るため、
①添付文書の届出義務の創設
②医療機器の登録認証機関による認証範囲の拡大
③再生医療等製品の条件及び期限付承認制度の創設等を内容とする改正法案
を、平成25年5月24日に第183回通常国会に提出し、継続審議となったが、平成26年11月20日の臨時国会で成立、11月27日公布された。
【施行日:公布日から1年以内の政令で定める日】
↓
2014年11月25日より改正法の施行が決定した。
医薬品医療機器等法の概要
1.医薬品、医療機器等に係る安全対策の強化
(1) 薬事法の目的に、保健衛生上の危害の発生・拡大防止のため必要な規制を行うことを明示する。
(2) 医薬品等の品質、有効性及び安全性の確保等に係る責務を関係者に課す。
(3) 医薬品等の製造販売業者は、最新の知見に基づき添付文書を作成し、厚生労働大臣に届け出るものとする。併せて、迅速な情報提供を行う観点から、届け出た添付文書を直ちにウェブサイトに掲載することとする。
2.医療機器の特性を踏まえた規制の構築
(1) 医療機器の製造販売業・製造業について、医薬品等と章を区分して規定する。
(2) 医療機器の民間の第三者機関による認証制度を、基準を定めて高度管理医療機器にも拡大する。
(3) 診断等に用いる単体プログラムについて、医療機器として製造販売の承認・認証等の対象とする。
(4) 医療機器の製造業について、許可制から登録制に簡素化する。
(5) 医療機器の製造・品質管理方法の基準適合性調査について、合理化を図る。
3.再生医療等製品の特性を踏まえた規制の構築
(1) 「再生医療等製品」を新たに定義するとともに、その特性を踏まえた安全対策等の規制を設ける。
(2) 均質でない再生医療等製品について、有効性が推定され、安全性が認められれば、特別に早期に、条件及び期限を付して製造販売承認を与えることを可能とする。
4.その他
薬事法の題名を「医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律」に改めるほか、所要の改正を行う。
認証制度の拡大
高度管理医療機器(クラスⅢ機器)にも認証制度を広げる。(現在はクラスⅢ、Ⅳは承認制)
これまでは、
・管理医療機器(クラスⅡ認証基準あり)は品目ごとに登録認証期間の認証を受け
・高度管理医療機器(クラスⅢ、クラスⅣ)は独立行政法人医薬品医療機器総合機構を経由して厚生労働大臣の承認を受けることとされていた。
法改正後は、高度管理医療機器のうち、後発品(市場で使用されている既存品と殆ど変わらない申請品)についても、認証制度が導入される。
ただし、法改正時に全品目が一気に認証制度に移行されるわけではなく、認証基準の制定がなされた品目から順に認証制度に移行されることになる。
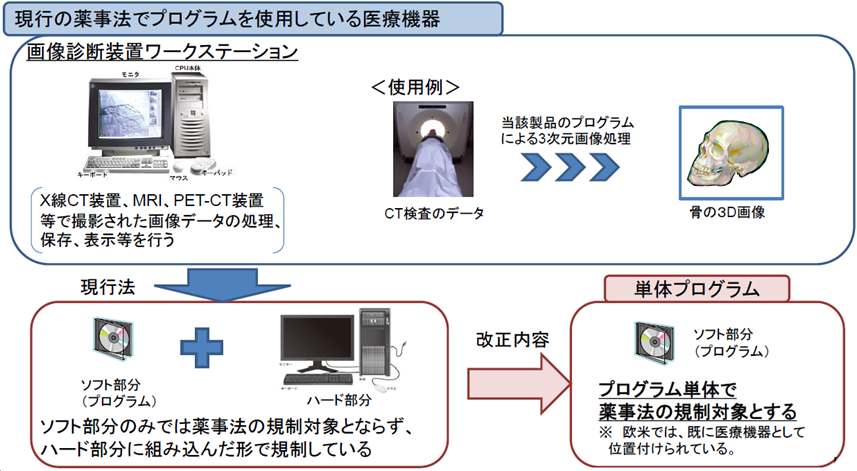
単体プログラムの医療機器化
これまでは、医療機器と併用して用いるコンピュータなどに用いられるソフトウェアのうち、現在はハードを伴わない単独のプログラム(画像診断装置等)は医療機器としては扱われて来なかった。
今後は「単独のプログラム」も医療機器として扱われるようになる。
(新たな法律では「電子計算機に対する指令であって、一の結果を得ることができるように組み合させれたもの、及びこれを記録した記録媒体」と規定されている。)
具体的にどのようなプログラムが医療機器として扱われるのかは現段階(平成26年6月現在)では明らかではないが、医療機器にかかわるプログラムを取り扱う業者は今後医療機器関連業者として取り扱われることになるので、製造販売業の許可、製造業の登録が必要となるため、注意が必要である。
規制の適用範囲に関しては現在検討中である。
第2条第1項第2号
人又は動物の疾病の診断、治療又は予防に使用されることが目的とされている物であつて、機械器具等(機械器具、歯科材料、医療用品、衛生用品並びにプログラム(電子計算機に対する指令であつて、一の結果を得ることができるように組み合わされたものをいう。以下同じ。)及びこれを記録した記録媒体をいう。以下同じ。)でないもの(医薬部外品及び再生医療等製品を除く。)
第2条第1項第13号
この法律で「製造販売」とは、その製造(他に委託して製造する場合を含み、他から委託を受けて製造をする場合を除く。以下「製造等」という。)をし、又は輸入をした医薬品(原薬たる医薬品を除く。)、医薬部外品、化粧品、医療機器若しくは再生医療等製品を、それぞれ販売し、貸与し、若しくは授与し、又は医療機器プログラム(医療機器のうちプログラムであるものをいう。以下同じ。)を電気通信回線を通じて提供することをいう。
「設計を行う者」が製造業者となる
医療機器の開発には「設計」がつきものである。医療機器の設計は、一般的には製造業者にて行われることが多いが、これまでは「設計」に対して許可が求められることはなかった。
新たな法律では、「設計」が製造業の一類型として位置づけられ、登録の対象となることになった。
このため、製造業者であっても、また、製造業者以外の設計業者であっても、別途、製造業の登録が必要となる。
製造業(外国製造業の認定制度)の登録制への移行
現在は製造業も認可制であるが、登録制に移行する。
これまで、医療機器の製造業を営むには医療機器製造業の「許可」が必要であった。この許可制度というのは、法令の定めた一定の要件を満たした業者が申請をし、審査をパスした場合にだけ、特別に許可が与えられるという制度である。
一方、登録制度は、許可制度とは異なり特別な許可は必要ではなく、一定の要件のもと、登録を受けることで事業を行える制度である(通常、更新は不要)。
今回の改正法では「構造設備基準」が適用されなくなる。
(製造業の登録)
第23条第2項第3号
業として、医療機器又は体外診断用医薬品の製造(設計を含む。以下この章及び第八十条第二項において同じ。)をしようとする者は、製造所(医療機器又は体外診断用医薬品の製造工程のうち設計、組立て、滅菌その他の厚生労働省令で定めるものをするものに限る。以下この章及び同項において同じ。)ごとに、厚生労働省令で定めるところにより、厚生労働大臣の登録を受けなければならない。
「製造」の概念の明確化
従来は、医療機器の「製造」の概念が明確でなく、単体で医療機器となるものを製造するかどうか(滅菌・保管表示を含む)で判断せざるを得なかった。
このことは、医療機器製造業許可の要不要、承認申請時の審査において様々なトラブルを生んできたという経緯がある。
今回の改正では「医療機器又は体外診断用医薬品の製造工程のうち、設計、組立て、滅菌その他厚生労働省令で定めるものをするものに限る」と記載され、省令の発出により、「製造」の概念がより明確になることが期待される。
(製造業の登録)
第23条第2項第3号
業として、医療機器又は体外診断用医薬品の製造(設計を含む。以下この章及び第八十条第二項において同じ。)をしようとする者は、製造所(医療機器又は体外診断用医薬品の製造工程のうち設計、組立て、滅菌その他の厚生労働省令で定めるものをするものに限る。以下この章及び同項において同じ。)ごとに、厚生労働省令で定めるところにより、厚生労働大臣の登録を受けなければならない。
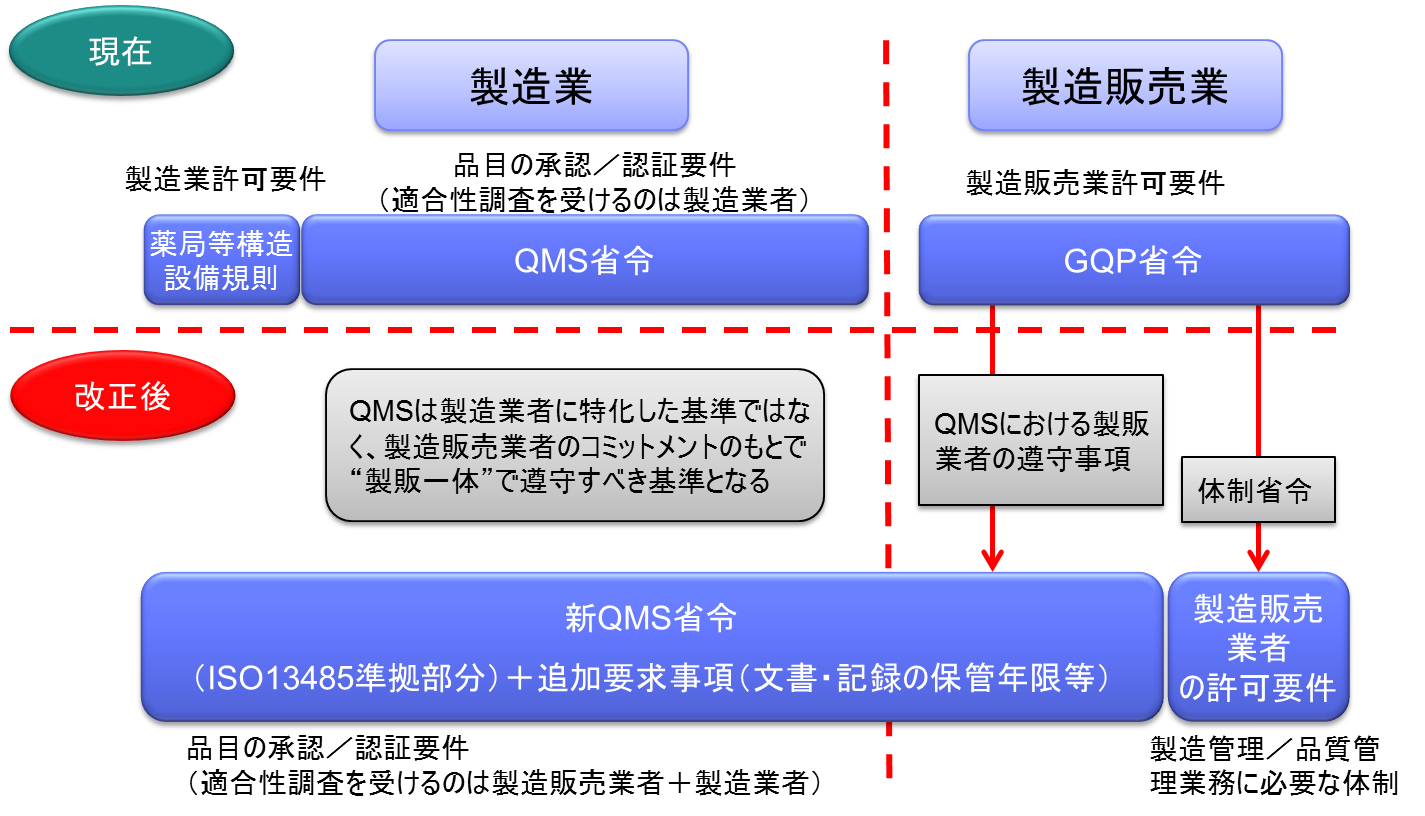
製造販売業がQMSの対象となる
これまでは、医療機器製造販売業の許可を受けるにあたって必要とされてきた要件は多々あったが、主なものはGVP(製造販売後安全管理体制)とGQP(品質管理体制)であった。
このうち、GQPとして要求されていた要件が「製造管理又は品質管理に係る業務を行う体制」に変わる。
これは、QMSと呼ばれているものである。
つまり、製造販売業の許可要件であるGQPがQMSに変更となる。
ただし、現行のQMS省令は製造販売業に対して適用されることを前提としていないため、製造販売業と製造業を包括した形の「新QMS省令」が発出される見込みである。
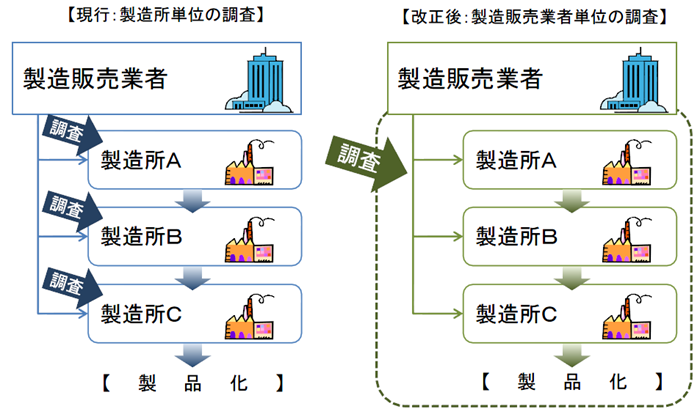
QMS調査の合理化
QMS(Quality Management System)調査の合理化
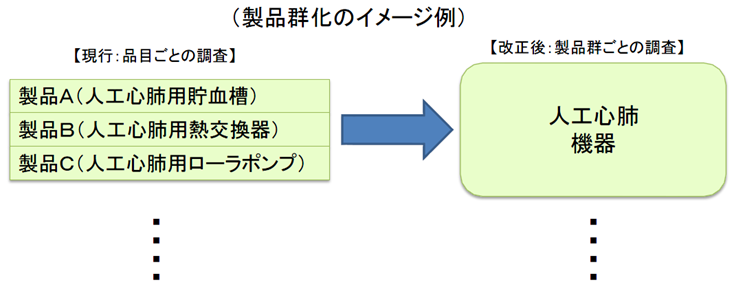
同一の製造販売業者において、既に製品AについてのQMS調査で基準に適合しているときは、製品B、CについてのQMS調査が原則免除される。
医薬品医療機器等法の施行にともなう政省令等の改正
医薬品医療機器等法の施行に向けて、
- 新法の「施行令」
- 新法の「施行規則」
- 「新QMS省令」
- 製造販売業者の許可基準たる「体制省令」
- 新法下におけるQMS調査の単位となる「製品群区分省令」
などがまとめて公布される予定。
各種運用通知等の検討も進められている。
医薬品医療機器等法のポイント(医療機器)
医療機器の製造販売業・製造業について、医薬品等と章を区分して規定する。
都道府県によるQMS調査の廃止
- 登録制+第三者認証
- 問題事案に対する立入検査等は都道府県が引き続き実施
「基準適合証」の新規導入
- 調査回数が減少
医療機器の分類と規制
医療機器の民間の第三者機関による認証制度を、基準を定めて高度管理医療機器にも拡大する。
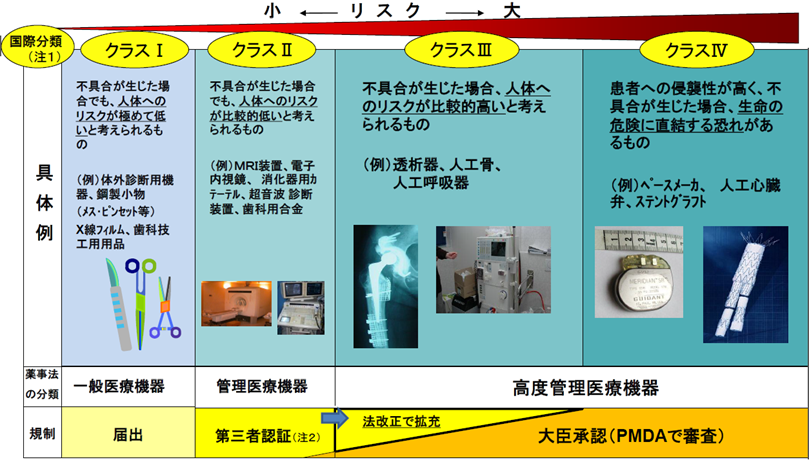
(注1) 日米欧豪加の5地域が参加する「医療機器規制国際整合化会合(GHTF)において平成15年12月に合意された医療機器のリスクに応じた4つのクラス分類の考え方を薬事法に取り入れている。
(注2) 厚生労働大臣が基準を定めたものについて大臣の承認を不要とし、あらかじめ厚生労働大臣の登録を受けた民間の第三者 認証機関(現在13機関)が基準への適合性を認証する制度。
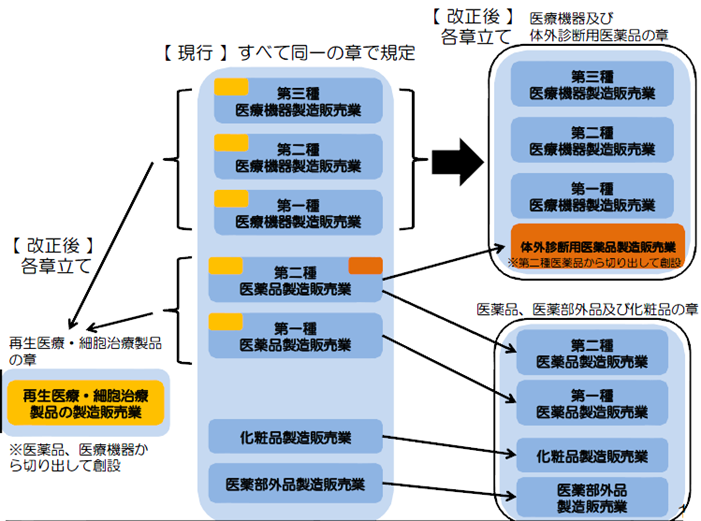
体外診断用医薬品が医療機器となる。
体外診断用医薬品製造販売業許可の取得が必要(第23条の2)
製造業の登録が必要(23条2の3)
医療機器等総括販売責任者:薬剤師または一部の体外診断用医薬品の場合は技術者で代用可(第23条の2の14)
体外診断用医薬品製造管理者:製造所毎に薬剤師または技術者(第23条2の14の5)
医療機器の製造業(外国製造業の認定制度)について、許可制から登録制に簡素化する。
医療機器の製造業(外国製造業の認定制度)について、許可制から登録制に簡素化する。
- 登録時の実地調査は、原則として不要。
- 登録の対象となる製造所は、現行に比して限定される。
- 登録制への移行に伴い、登録申請時に添付する資料を簡素化。
【現行】すべて許可制 【改正後】医療機器、体外診断用医薬品は登録制へ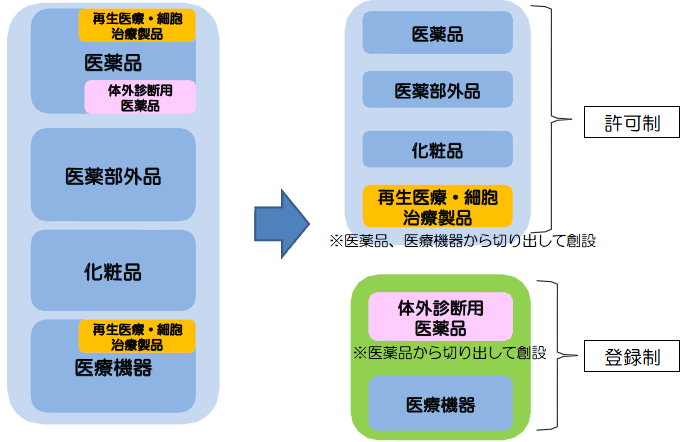
医療機器の「機械器具等」の範疇に、「ソフトウェア(プログラム)」を追加。
診断等に用いる単体プログラムについて、医療機器として製造販売の承認・認証等の対象とする。
医療機器とされるソフトウェアを設計・開発・製造する業者も製造業の登録が必要。
単体プログラム 汎用PC等にインストールすることで、医療機器としての性能を発揮するプログラム※
※プログラム・・・電子計算機に対する指令であって一の結果を得ることができるように組み合わされたもの
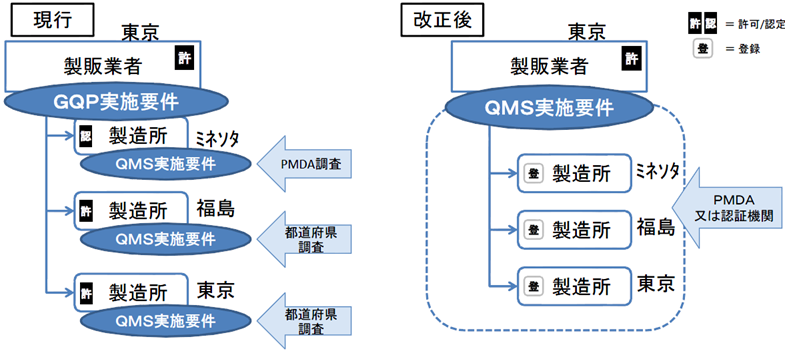
医療機器の製造・品質管理方法の基準適合性調査について、合理化を図る。
製造所毎に別個に調査・判定をするのではなく、製造販売業者に対して、システム全体を包括的に調査・判定。
製造販売業がQMS省令の対象となる。
これまでは個々の製造所ごとにその品質管理要件としてQMS適合性を求めてきた。
医療機器は、複数の製造所を含むシステムとしての品質管理が重要であるため、全製造所を統括したQMSとして管理すべきであり、全体のQMS適合性を製販業者に対する要件とする。
QMSは広域的なひとつの品質管理システムとなるため、一製造所ごとの所管都道府県による調査を廃止し、全体をひとつとして調査・評価できるよう、広域的な機関として、PMDAまたは登録認証機関に調査権を集約する。
製造販売業の許可の基準として「QMS」が遵守要件となる
製造所毎に別個に調査・判定をするのではなく、製造販売業者に対して、システム全体を包括的に調査・判定。
「製造」の概念の明確化。
現行では許可の対象とされていない「設計を行う製造所」が、概念として追加される。
その他
関係条例の整備(手数料の設定等)
都道府県のシステムの改修

{kind=link}
I pay a visit everyday some web sites and blogs to read content, but this webpage gives quality based articles.
What i don’t understood is in fact how you are no longer actually much more neatly-appreciated than you might be right
now. You are so intelligent. You already know thus significantly on the subject of this matter, made me individually believe
it from a lot of numerous angles. Its like women and men aren’t fascinated until
it is one thing to accomplish with Woman gaga! Your individual stuffs outstanding.
Always take care of it up!