
製造業者は機器のMDRの適合を行う場合、適合性評価手順(Conformity Assessment Procedures)に基づく必要がある。…
製造業者は、医療機器のリスクに応じた「適合性評価手順」を見極める必要がある。
適合性評価手順はArticle 52 「適合性評価手順」に記載されている。
「製造業者は、機器を上市する前に、当該機器の適合性評価を、Annex Ⅸ~Ⅺ に規定されている該当する適合性評価手順に従って実施しなければならない。」
適合性評価手順は機器のクラス等により異なる。
- 日本、米国では、行政当局(または権限を委託された外部機関)が新しい医療機器について市販前の承認(認証)を与えるというシステムを採用 l製造業者の観点から見ると、医療機器の市販前に審査を受け、その結果として「承認(認証)を与えられる」形となる(受動形)
- 欧州では製造業者が自ら規制要求事項への適合を「宣言」し、CEマーキングを表示するという考え方を採用(能動形)
機器のクラス分類がClass Is,Ir,ImまたはClass IIa以上の場合、製造業者が適合宣言をするためのプロセスの中でNBの適合性評価が必要
そのため、製造業者が完全に自らの意思のみで宣言できるわけではない。
しかしながら、最後は自ら宣言をして、製造業者の法的責任の下でCEマーキングを表示するのが欧州の考え方である。
そのため、CEマーキングは「取得する」という表現は使用しない。 「添付する」または「表示する」を使用する。
MDDと異なった適合性評価手順は以下のとおりである。
- Class IIb埋め込み医療機器
- クラスⅢとほぼ同じ扱いとなる。
- ただし、縫合糸、ステープル、歯科用充填物、歯列矯正具、歯冠材、スクリュー、くさび、プレート、ワイヤ、ピン、クリップ、コネクタは除く
- ClassⅠ再使用可能な外科用器具
- クラスⅠ滅菌医療機器、測定機能を持つ医療機器とほぼ同じ扱いとなる
機器により、以下の特別な追加適合性手順がある。
- 医薬品を含む医療機器の場合、Annex Ⅸ Section 5.2またはAnnex X Section 6も適用すること。
所轄官庁(CA)または欧州医薬品庁(EMA)の評価がある。 - ヒト血液由来を含む機器は、出荷前にバッチ検証が必要。
- ヒト組織または細胞由来を含む医療機器の場合、Annex Ⅸ Section 5.3またはAnnex X Section 6も適用すること。
所轄官庁(CA)の評価がある。 - 人体開口部を経てまたは皮膚に適用され、人体に入れられ、人体内で吸収または局所的に分散されることを意図した物質、または物質のコンビネーション医療機器は、Annex Ⅸ Section 5.4 またはAnnex X Section 6も適用すること。
所轄官庁(CA)または欧州医薬品庁(EMA)の評価がある。
適合性評価手順(Article 52)
- 製造業者は、機器を上市する前に、当該機器の適合性評価を、Annex Ⅸ~Ⅺに規定されている該当する適合性評価手順に従って実施しなければならない。
- 製造業者は、上市されていない機器の使用を開始する前に、当該機器の適合性評価を、Annex Ⅸ~Ⅺに規定されている該当する適合性評価手順に従って実施しなければならない。
- クラスⅢの機器(ただし、カスタムメイド機器または治験機器は除く)の製造業者は、Annex Ⅸに定められているように、適合性評価の対象とならなければならない。あるいは、当該製造業者は、Annex Xに定められている適合性評価とAnnex Ⅺに定められている適合性評価を併せて適用することを選択してもよい。
- クラスⅡb機器(ただし、カスタムメイド機器または治験機器は除く)の製造業者は、Annex Ⅸの第I章および第Ⅲ章に定められているように、適合性評価の対象とならなければならない。これには、ジェネリック機器グループごとに少なくとも一つの代表機器に関する、当該Annexの第4節に定められている技術文書の評価を含める。ただし、縫合糸、ステープル、歯科用充填剤、歯列矯正具、歯冠、ねじ、くさび、プレート、ワイヤ、ピン、クリップおよびコネクタを除くクラスⅡb埋込機器については、Annex Ⅸの第4節に定められているように、技術文書の評価は、全ての機器に適用しなければならない。あるいは、当該製造業者は、Annex Xに定められている型式審査に基づく適合性評価とAnnex Ⅺに定められている製品適合性検証に基づく適合性評価を併せて適用すること選択してもよい。
- 本条項の第4項の中段に挙げた免除機器に使用されている、確立した技術に類似した技術がその他のクラスⅡb埋込機器に使用される正当性が認められた場合、または患者、使用者などの健康と安全若しくはその他の公衆衛生面を保護する正当性が認められた場合、欧州委員会は、その他のクラスⅡb埋込機器のリストヘの追加、または機器のリストからの削除といった修正を行うために、Article 115に従って委任法令を採択する権限が与えられる。
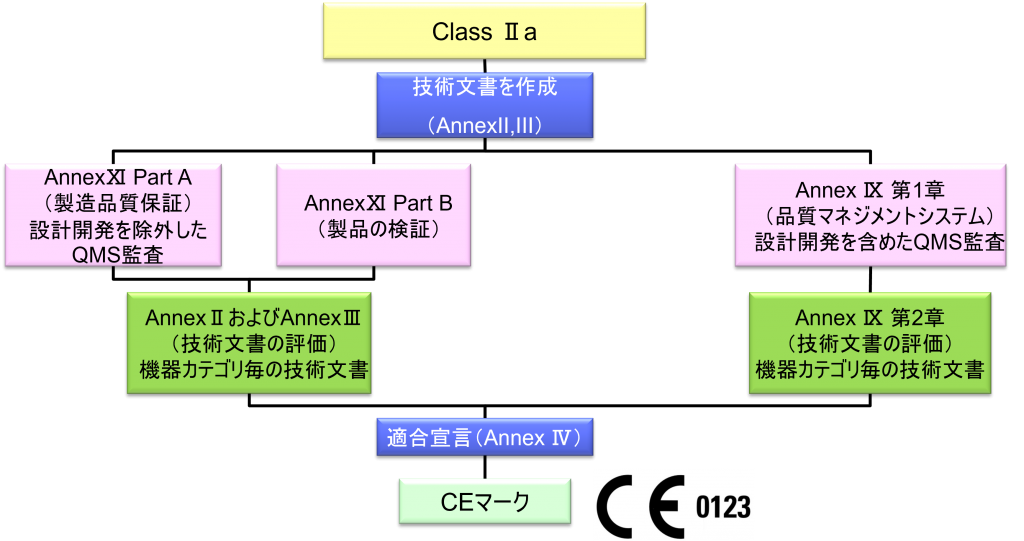
- クラスⅡa機器(ただし、カスタムメイド機器または治験機器は除く)の製造業者は、Annex Ⅸの第I章および第Ⅲ章に定められているように、適合性評価の対象とならなければならない。これには、機器のカテゴリーごとに少なくとも一つの代表機器に関する、当該Annex第4節に定められている技術文書の評価を含める。あるいは、当該製造業者は、Annex Ⅺの第10節または第18節に定められている適合性評価と併せて、AnnexⅡおよびⅢに規定されている技術文書を作成することを選択してもよい。技術文書の評価は、機器のカテゴリーごとに少なくとも一つの代表機器に対して適用しなければならない。
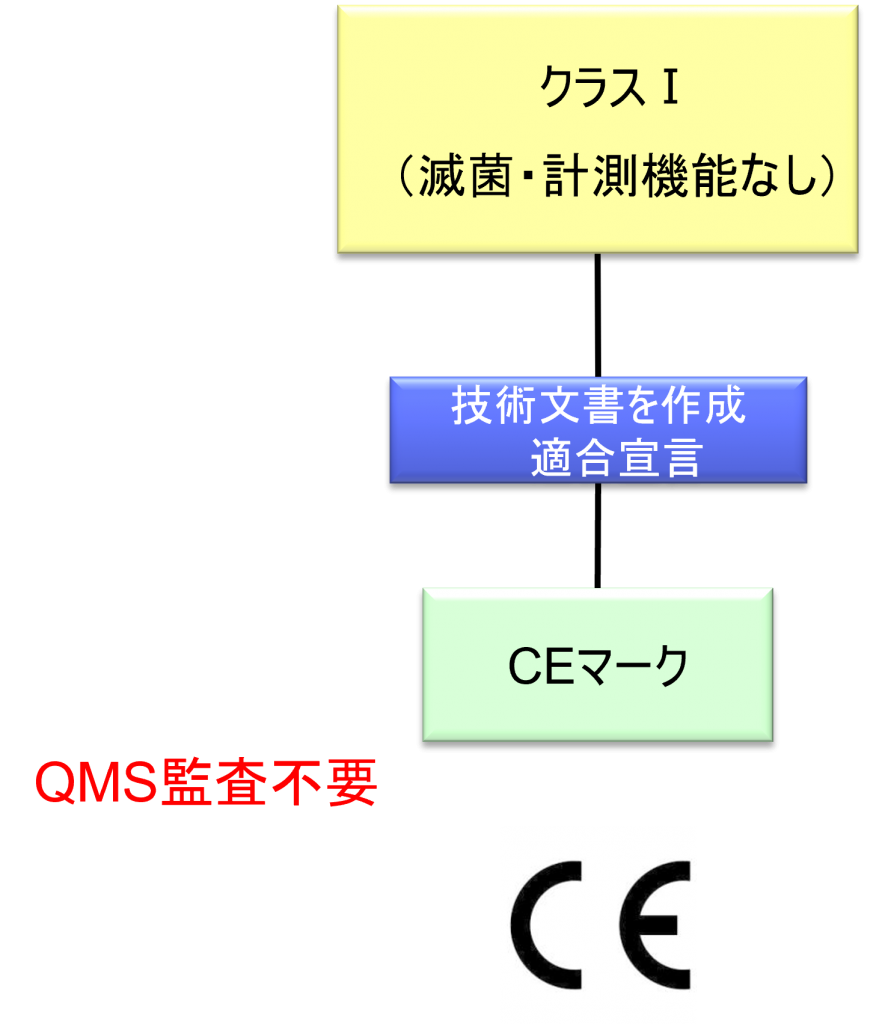
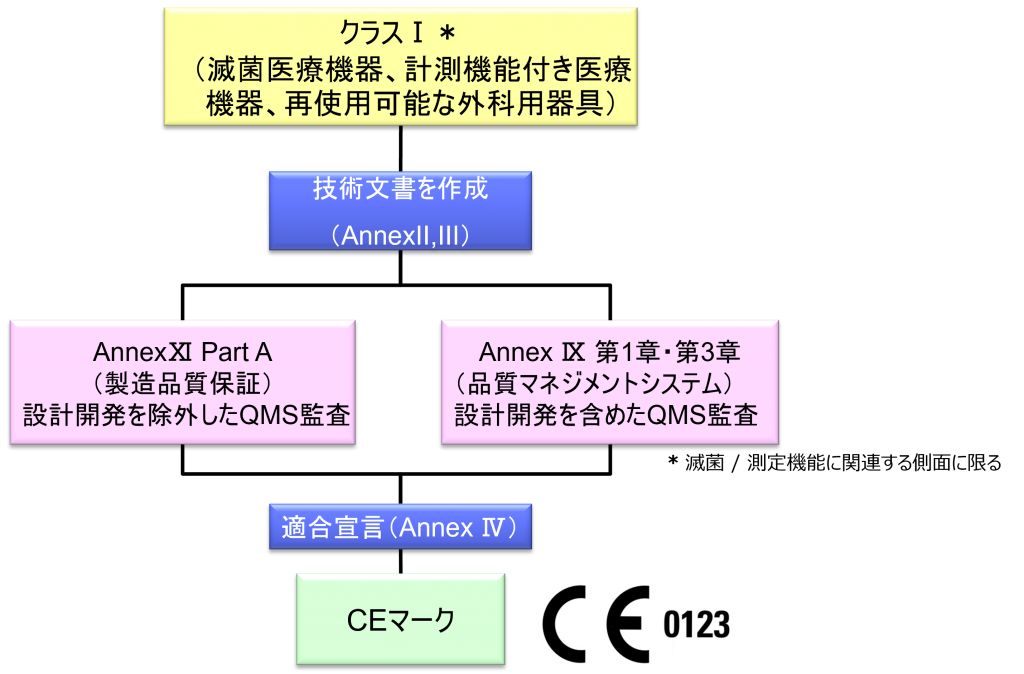
- クラスI機器(ただし、カスタムメイド機器または治験機器は除く)の製造業者は、AnnexⅡおよびⅢに規定されている技術文書の作成後、Article 19に規定されているEU適合宣言書を発行して製品の適合性を宣言しなければならない。それらの機器が滅菌状態で上市される、計測機能を有する、または再使用可能な外科用器具である場合、製造業者は、Annex Ⅸの第I章および第Ⅲ章またはAnnex ⅪのパートAに規定されている手順を適用しなければならない。ただし、次の事項については、本手順におけるNBの関与は制限されなければならない。
a.滅菌状態で上市される機器の場合、滅菌条件の設定、確保および維持に関連する事項
b.計測機能を有する機器の場合、計量要件への適合性に関する事項
c.再使用可能な外科用器具の場合、機器の再使用、特に洗浄、消毒、滅菌、保守および機能検査、並びに該当する取扱説明書に関する事項 - カスタムメイド機器の製造業者は、Annex XIIIに規定されている手順に従い、そうした機器を上市する前に、当該Annexの第1節に規定されているステートメントを作成しなければならない。前段に準拠して適用される手順に加え、クラスⅢカスタムメイド埋込機器の製造業者は、Annex Ⅸの第1章に定められている適合性評価手順の対象とならなければならない。あるいは、当該製造業者はAnnex ⅪのパートAに定められている適合性評価の適用を選択してもよい。
- Article 1第8項の前段に規定されている機器の場合、本条項の第3、4、6または7項に準拠して適用される手順に加え、適用対象となる場合は、Annex Ⅸの第5.2節またはAnnex Xの第6節に規定する手順も適用しなければならない。
- Article 1第6項の(f)または(g)およびArticle 1第10項の前段に従って、本規制の対象である機器の場合、本条項の第3、4、6または7項に準拠して適用される手順に加え、適用対象となる場合は、Annex Ⅸの第5.3節またはAnnex Xの第6節に規定する手順も適用しなければならない。
- 人体開口部を介して人体へ入れる、または皮膚に塗布することが意図され、人体に吸収される、または局所的に分散される物質若しくは物質の組み合わせから成る機器の場合、第3、4、6または7項に準拠して適用される手順に加え、適用対象となる場合は、Annex Ⅸの第5.4節またはAnnex Xの第6節に定められている手順も適用しなければならない。
- NBが設立されている加盟国は、第1項~第7項および第9項~第11項に規定されている手順に関する全ての文書または特定の文書(技術文書、監査、評価および検査の報告書を含む)が、加盟国が決定した欧州連合の公用語版で入手できるよう要求してもよい。そのような要求事項を定めない場合、それらの文書は、NBの同意する欧州連合の公用語で提供されなければならない。
- 治験機器は、Article 62~Article 81に規定されている要求事項の対象としなければならない。
- 欧州委員会は、NBによる適合性評価手順の統一的な適用を保証することを目的として、実施法令により次の点に関する詳細な取り決めおよび手順を規定してもよい。
a.クラスⅡaおよびクラスⅡbの機器については、Annex Ⅸの第2.3節第3項および第3.5 節に、クラスⅡaの機器については、Annex Ⅺの第10.2節に規定されている代表機器の技術文書の評価の頻度およびサンプリング
b.機器のリスクのクラスおよび種類を考慮し、Annex Ⅸの第3.4節に従ってNBが実施する予告なしの現地監査および抜取試験の最低頻度
c.Annex Ⅸの第3.4節および第4.3節、Annex Xの第3節、並びにAnnex Ⅺの第15節に従った抜取試験、技術文書の評価および型式審査においてNBが実施する物理的試験、臨床試験またはその他の試験
前段に規定された当該実施法令は、Article 114第3項に規定されている審査手順に従って採択されなければならない。
クラスⅠ機器(滅菌・計測機能なし)の適合性評価ルート
クラスI*(滅菌医療機器、計測機能付き医療機器、再使用可能な外科用器具)機器の適合性評価ルート
Class Ⅱa
ClassⅡb (能動医療機器で医薬品を投与または除去するもの)
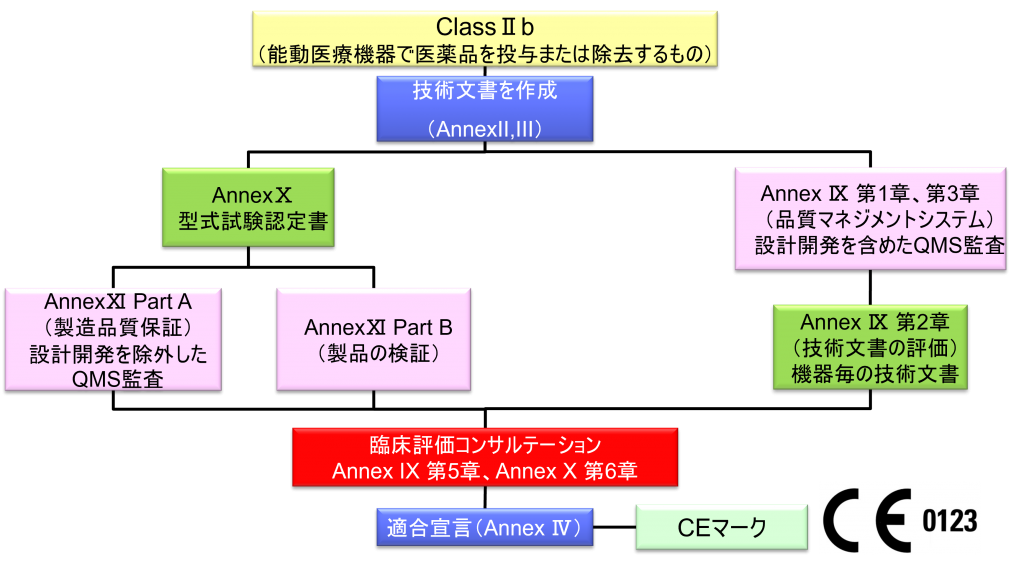
ClassⅡb(非埋め込み医療機器)
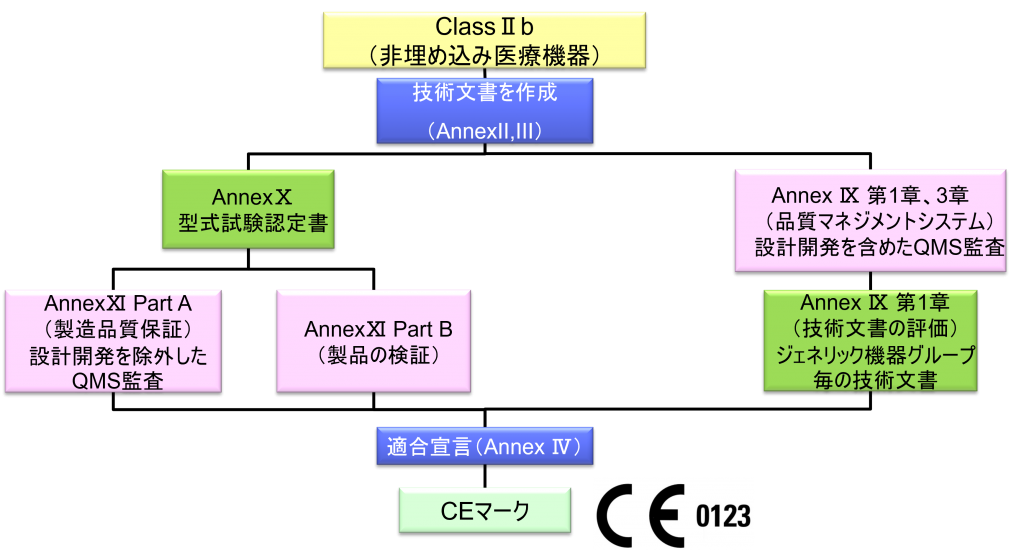
ClassⅡb(埋め込み医療機器)WET(Well-established Technology)
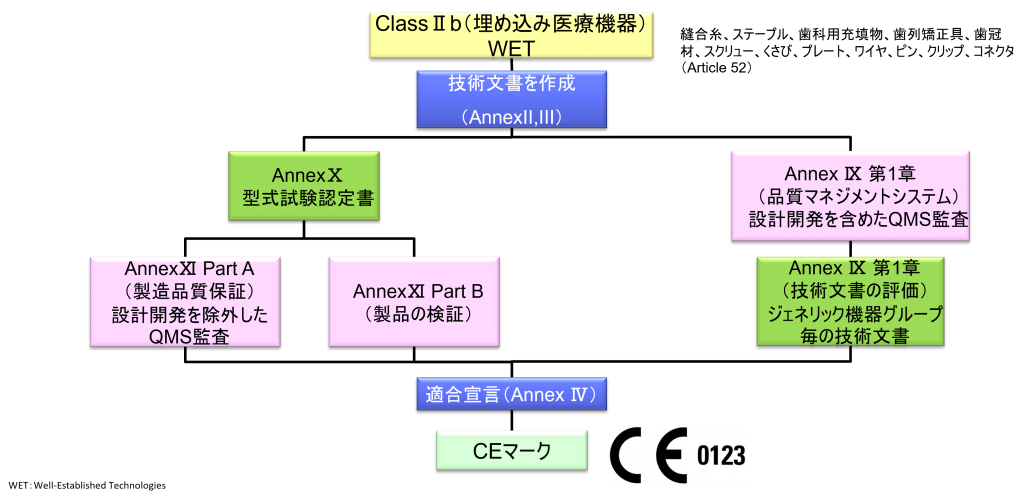
ClassⅡb(埋め込み医療機器)non-WET

Class Ⅲ非埋込医療機器の適合性評価ルート
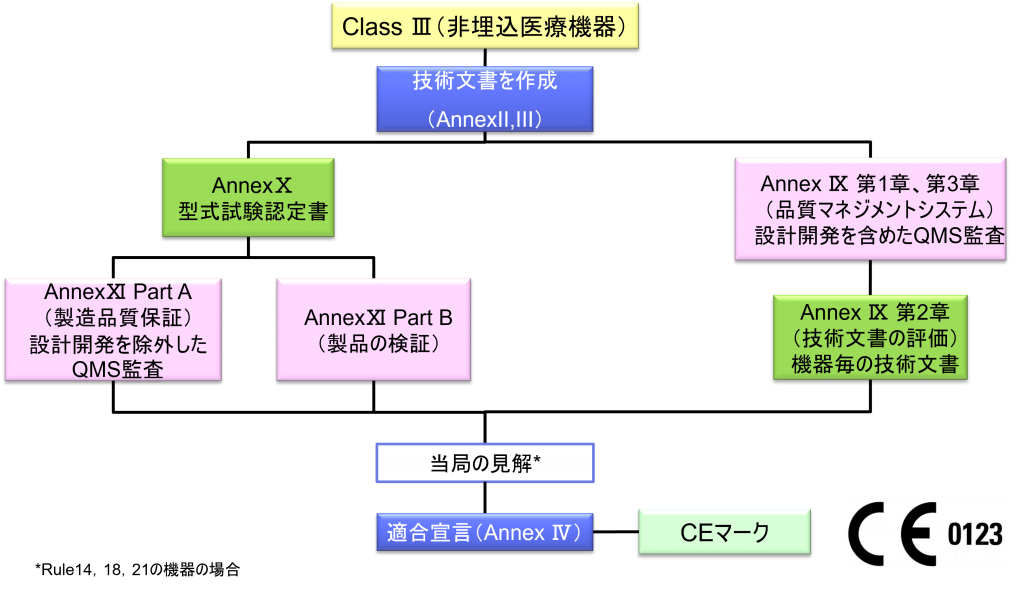
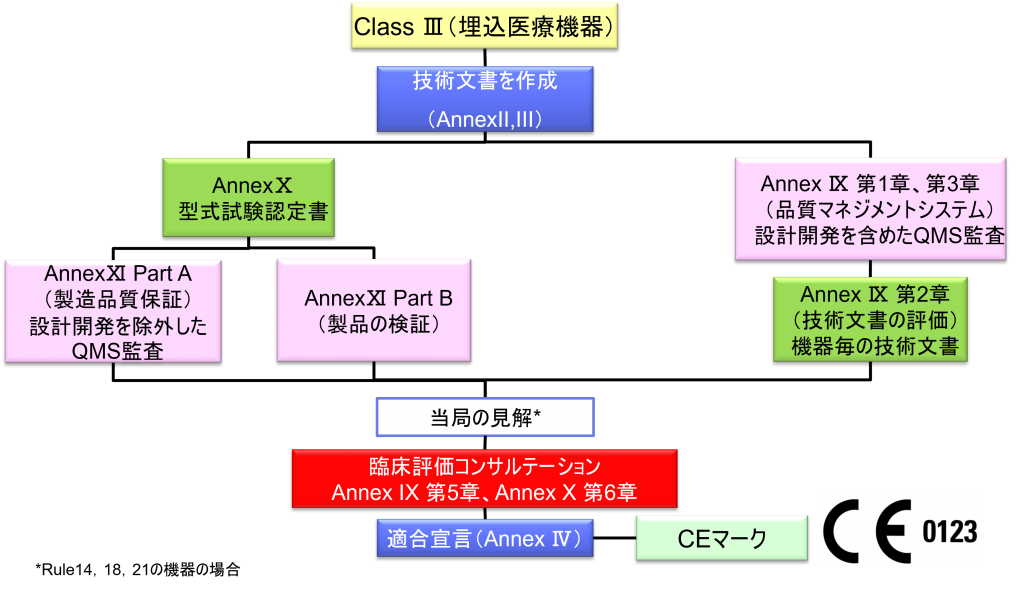
Class Ⅲ埋込医療機器の適合性評価ルート
お役立ち動画
世界一わかりやすいMDRセミナー
【第1講】欧州制度概要
【第2講】MDR概要
【第3講】規則実行に関連する機関等
【第4講】移行スケジュール
【第5講】MDRの要点
【第6講】用語の定義
【第7講】概要製造業者の責務
【第8講】規制遵守責任者
【第9講】一般的なMDR対応の流れ
【第10講】MDRが適用される機器・製品
【第11講】クラス分類
【第12講】安全性および性能の要求事項
【第13講】技術文書
【第14講】臨床評価
【第15講】 適合性評価手順
【第16講】欧州指定代理人
【第17講】輸入業者
【第18講】販売業者
【第19講】UDI
【第20講】EUDAMEDへの登録
関連商品
[blogcard url=”https://ecompliance.co.jp/SHOP/O063.html” title=”MDR(欧州医療機器規則)施行直前セミナー” content=”いよいよ5月26日にMDR(欧州医療機器規則)が完全施行されます。品目認証の有効期間に関わらず、5月26日以降はPMSおよびビジランスはMDRの要求を満たす必要があります。
また、クラスⅠ機器でNBの関与を必要としない機器に関しては、2021年5月26日からMDRを遵守する必要があります。
しかしながら、MDRは行政側の準備が整っていない面もあります。
たとえば、経済事業者の情報、UDI情報、安全性情報等を登録するためのデータベースであるEUDAMEDはまだ完全稼働していません。
この点に関しては、MDCGがMDCG 2021-1「EUDAMEDが完全に機能するまでの、調和のとれた管理慣行と代替技術ソリューションに関するガイダンス」(Guidance on harmonised administrative practices and alternative technical solutions until EUDAMED is fully functional)を発出しています。
EUDAMEDが稼働するまでの代替プロセスが解説されています。
本セミナーでは、MDR完全施行に伴い、医療機器企業が準備しなければならない事項を分かりやすく解説します。”]
[blogcard url=”https://ecompliance.co.jp/SHOP/EL-080.html” title=”【セミナービデオ】MDR(欧州医療機器規則)施行直前セミナー” content=”いよいよ5月26日にMDR(欧州医療機器規則)が完全施行されます。品目認証の有効期間に関わらず、5月26日以降はPMSおよびビジランスはMDRの要求を満たす必要があります。
また、クラスⅠ機器でNBの関与を必要としない機器に関しては、2021年5月26日からMDRを遵守する必要があります。
しかしながら、MDRは行政側の準備が整っていない面もあります。
たとえば、経済事業者の情報、UDI情報、安全性情報等を登録するためのデータベースであるEUDAMEDはまだ完全稼働していません。
この点に関しては、MDCGがMDCG 2021-1「EUDAMEDが完全に機能するまでの、調和のとれた管理慣行と代替技術ソリューションに関するガイダンス」(Guidance on harmonised administrative practices and alternative technical solutions until EUDAMED is fully functional)を発出しています。
EUDAMEDが稼働するまでの代替プロセスが解説されています。
本セミナーでは、MDR完全施行に伴い、医療機器企業が準備しなければならない事項を分かりやすく解説します。”]
[blogcard url=”https://ecompliance.co.jp/SHOP/L_MDR.html” title=”欧州医療機器規則MDR(Medical Device Regulation)セミナー” content=”2017年5月5日に、医療機器指令 Medical Device Directive (93/42/EEC)が改正され 「欧州医療機器規則 (MDR:Medical Device Regulation)」 が公示されました。20日後の2017年5月25日より施行され、2020年5月25日までの3年間が移行措置期間とされています。Manufacturer は、この移行期間中に技術文書を改訂し、新しい要求事項に対応しなければなりません。
MDRは、従来のMDDと能動埋め込み型医療機器指令 Active Implantable Medical Device Directive (90/385/EEC)を統合し置き換えるものです。
MDDからMDRへの改正内容は多岐に渡り、また上市済の製品の対策も必要となるため、強制化に間に合うように計画的に対策を進める必要があります。
MDRは、日本や米国の法規制を参考にして作成され、世界で最も厳しい規制となりました。
しかしながらMDRは条文が難解で非常にわかりにくいのが難点です。
いったい何が要求されどのように対応したら良いのでしょうか?
MDRは、技術文書について、従来よりも踏み込んだ精査を求めています。
また臨床評価と市販後臨床フォローアップについてより厳しい要求事項となりました。
本セミナーでは、MDRの要点をわかりやすく解説し、MDDからの変更点を明らかにします。また優先すべき事項、対応のための問題点・リスクを解説します。”]
[blogcard url=”https://ecompliance.co.jp/SHOP/EL-028.html” title=”【セミナービデオ】欧州医療機器規則MDR(Medical Device Regulation)セミナー” content=”2017年5月5日に、医療機器指令 Medical Device Directive (93/42/EEC)が改正され 「欧州医療機器規則 (MDR:Medical Device Regulation)」 が公示されました。
20日後の2017年5月25日より施行され、2020年5月25日までの3年間が移行措置期間とされています。Manufacturer は、この移行期間中に技術文書を改訂し、新しい要求事項に対応しなければなりません。
MDRは、従来のMDDと能動埋め込み型医療機器指令 Active Implantable Medical Device Directive (90/385/EEC)を統合し置き換えるものです。
MDDからMDRへの改正内容は多岐に渡り、また上市済の製品の対策も必要となるため、強制化に間に合うように計画的に対策を進める必要があります。
MDRは、日本や米国の法規制を参考にして作成され、世界で最も厳しい規制となりました。
しかしながらMDRは条文が難解で非常にわかりにくいのが難点です。
いったい何が要求されどのように対応したら良いのでしょうか?
MDRは、技術文書について、従来よりも踏み込んだ精査を求めています。
また臨床評価と市販後臨床フォローアップについてより厳しい要求事項となりました。
本セミナーでは、MDRの要点をわかりやすく解説し、MDDからの変更点を明らかにします。また優先すべき事項、対応のための問題点・リスクを解説します。”]
[blogcard url=”https://ecompliance.co.jp/SHOP/200716-P.html” title=”欧州医療機器規則(MDR)におけるPMS・ビジランス対応セミナー” content=”コロナ禍によりMDR(Medical Device Regulation:欧州医療機器規則)の完全施行が2021年5月まで延期となりました。日本の企業はMDRの全貌を適切に理解していないと思われ、対応が後手に回っていると思われます。
MDRは、FDAや日本の規制要件、および医薬品の規制要件などを参考に構築されており、いわば世界一厳しい医療機器規制要件となりました。
とりわけ、市販後監視(PMS)およびビジランスシステムに関しては難解であり、適切に理解している企業は稀であると言っても過言ではないでしょう。
本邦においては、QMS省令とGVP省令は明確に区別されていますが、MDRにおいては、PMS、ビジランスの要求事項もQMSに取り入れて構築する必要があります。
また当該QMSは欧州の手順のみを記載すれば良いのではなく、日本や米国など欧州圏外のPMSやビジランスの手順をすべて含めることとされています。
また市販後監視に関わる技術文書の要求事項が新設され、市販後監視計画書なども技術文書として管理・維持することが求められています。
本セミナーでは、医薬品GVPを熟知した講師が、欧州医療機器規則におけるPMSおよびビジランスに関して分かりやすく解説を行います。”]
[blogcard url=”https://ecompliance.co.jp/SHOP/EL-039.html” title=”【セミナービデオ】欧州医療機器規則(MDR)におけるPMS・ビジランス対応セミナー” content=”コロナ禍によりMDR(Medical Device Regulation:欧州医療機器規則)の完全施行が2021年5月まで延期となりました。日本の企業はMDRの全貌を適切に理解していないと思われ、対応が後手に回っていると思われます。
MDRは、FDAや日本の規制要件、および医薬品の規制要件などを参考に構築されており、いわば世界一厳しい医療機器規制要件となりました。
とりわけ、市販後監視(PMS)およびビジランスシステムに関しては難解であり、適切に理解している企業は稀であると言っても過言ではないでしょう。
本邦においては、QMS省令とGVP省令は明確に区別されていますが、MDRにおいては、PMS、ビジランスの要求事項もQMSに取り入れて構築する必要があります。
また当該QMSは欧州の手順のみを記載すれば良いのではなく、日本や米国など欧州圏外のPMSやビジランスの手順をすべて含めることとされています。
また市販後監視に関わる技術文書の要求事項が新設され、市販後監視計画書なども技術文書として管理・維持することが求められています。
本セミナーでは、医薬品GVPを熟知した講師が、欧州医療機器規則におけるPMSおよびビジランスに関して分かりやすく解説を行います。”]
[blogcard url=”https://ecompliance.co.jp/SHOP/200729-P.html” title=”欧州医療機器規則(MDR)における臨床評価セミナー” content=”コロナ禍によりMDR(Medical Device Regulation:欧州医療機器規則)の完全施行が2021年5月まで延期となりました。日本の企業はMDRの全貌を適切に理解していないと思われ、対応が後手に回っていると思われます。
MDRは、FDAや日本の規制要件、および医薬品の規制要件などを参考に構築されており、いわば世界一厳しい医療機器規制要件となりました。
とりわけ、MDRでは臨床評価要求事項(臨床的な安全評価・有効性評価)が厳格化されました。
臨床評価はMDD(Annex X)でも要求されていましたが、MDRにおいても臨床評価を実施し、臨床評価報告書を作成する必要があります。
臨床評価は機器クラスや新規性とは関係なく、全ての機器に必要であり、日本の要求と異なることの1つです。
実施のための詳細は臨床評価ガイダンス(MEDDEV 2.7/1 revision 4)に記載されています。
MDRの臨床評価では、ガイダンスの内容の大項目の一部を含めた上で、さらに要件の追加および変更を行っています。
臨床データと臨床評価報告書(CER)は精査し、また、これらを繰り返し更新することも要求されます。
製造業者は、MDR Article 61およびAnnex XIVにPart A(臨床評価関連のAnnex)に基づいて臨床評価と市販後臨床フォローアップ(PMCF)を実施しなければなりません。
製造業者は臨床評価レベルの適切性について正当化することが求められます。
臨床評価は下記に基づく必要があります。
a)科学的文献(Scientific Literature)
b)臨床試験(Clinical Investigation)
c)他のオプションとなる治療法の考慮(Other treatment)
当然のことながら、認証機関の審査する技術資料の1つとして臨床評価が含まれます。
臨床評価コンサルテーションも導入され、リスクの高い医療機器について、臨床評価の審査に行政当局が介入することになりました。
それに伴い、臨床試験実施可否判断基準も厳格化されました。
MDRにおいてはリスクの高い医療機器は、臨床試験が原則的に必須です。
リスクの高い医療機器は、他社の医療機器との同等性を示す場合、その医療機器の技術文書へのフルアクセスの契約があることが条件となりますが、事実上は難しいでしょう。
埋め込み機器およびクラス?機器の場合は、安全性と臨床性能のサマリー(SSCP:Summary of Safety and Clinical Performance)の作成が求められます。
臨床評価は市販後調査(Post-Market Surveillance)および市販後臨床フォローアップ(Post-Market Clinical Follow-up)とも関連します。
市販後監視で集めた情報によって臨床評価を更新することも忘れてはなりません。
PMCFのより詳細な考察や、定期的な安全性アップデート報告書(クラス?a以上の機器)の提出が求められます。
本セミナーでは、日米欧の医療機器規制要件を熟知した講師が、欧州医療機器規則における臨床評価に関して分かりやすく解説を行います。”]
[blogcard url=”https://ecompliance.co.jp/SHOP/EL-040.html” title=”【セミナービデオ】欧州医療機器規則(MDR)における臨床評価セミナー” content=”コロナ禍によりMDR(Medical Device Regulation:欧州医療機器規則)の完全施行が2021年5月まで延期となりました。日本の企業はMDRの全貌を適切に理解していないと思われ、対応が後手に回っていると思われます。
MDRは、FDAや日本の規制要件、および医薬品の規制要件などを参考に構築されており、いわば世界一厳しい医療機器規制要件となりました。
とりわけ、MDRでは臨床評価要求事項(臨床的な安全評価・有効性評価)が厳格化されました。
臨床評価はMDD(Annex X)でも要求されていましたが、MDRにおいても臨床評価を実施し、臨床評価報告書を作成する必要があります。
臨床評価は機器クラスや新規性とは関係なく、全ての機器に必要であり、日本の要求と異なることの1つです。
実施のための詳細は臨床評価ガイダンス(MEDDEV 2.7/1 revision 4)に記載されています。
MDRの臨床評価では、ガイダンスの内容の大項目の一部を含めた上で、さらに要件の追加および変更を行っています。
臨床データと臨床評価報告書(CER)は精査し、また、これらを繰り返し更新することも要求されます。
製造業者は、MDR Article 61およびAnnex XIVにPart A(臨床評価関連のAnnex)に基づいて臨床評価と市販後臨床フォローアップ(PMCF)を実施しなければなりません。
製造業者は臨床評価レベルの適切性について正当化することが求められます。
臨床評価は下記に基づく必要があります。
a)科学的文献(Scientific Literature)
b)臨床試験(Clinical Investigation)
c)他のオプションとなる治療法の考慮(Other treatment)
当然のことながら、認証機関の審査する技術資料の1つとして臨床評価が含まれます。
臨床評価コンサルテーションも導入され、リスクの高い医療機器について、臨床評価の審査に行政当局が介入することになりました。
それに伴い、臨床試験実施可否判断基準も厳格化されました。
MDRにおいてはリスクの高い医療機器は、臨床試験が原則的に必須です。
リスクの高い医療機器は、他社の医療機器との同等性を示す場合、その医療機器の技術文書へのフルアクセスの契約があることが条件となりますが、事実上は難しいでしょう。
埋め込み機器およびクラス?機器の場合は、安全性と臨床性能のサマリー(SSCP:Summary of Safety and Clinical Performance)の作成が求められます。
臨床評価は市販後調査(Post-Market Surveillance)および市販後臨床フォローアップ(Post-Market Clinical Follow-up)とも関連します。
市販後監視で集めた情報によって臨床評価を更新することも忘れてはなりません。
PMCFのより詳細な考察や、定期的な安全性アップデート報告書(クラス?a以上の機器)の提出が求められます。
本セミナーでは、日米欧の医療機器規制要件を熟知した講師が、欧州医療機器規則における臨床評価に関して分かりやすく解説を行います。”]
[blogcard url=”https://ecompliance.co.jp/SHOP/MD-QMS-169.html” title=”【MDR対応QMSひな形】サプライチェーン管理手順書” content=”CEマーキングを貼付し、欧州に医療機器を輸出している医療機器企業は、MDDに置き換わって2021年5月25日までに欧州医療機器規則(MDR:Medical Device Regulation)に対応する必要があります。製造業者(Manufacturer)は、この移行期間中に技術文書を改訂し、新しい要求事項に対応しなければなりません。
MDRは、従来のMDDと能動埋め込み型医療機器指令 Active Implantable Medical Device Directive (90/385/EEC)を統合し置き換えるものです。
MDDからMDRへの改正内容は多岐に渡り、また上市済の製品の対策も必要となるため、強制化に間に合うように計画的に対策を進める必要があります。
MDRは、日本や米国の法規制を参考にして作成され、世界で最も厳しい規制となりました。
しかしながらMDRは条文が難解で非常にわかりにくいのが難点です。
本邦におけるQMS省令や米国FDAのQSRにはない要求事項も含まれます。
そのため、多くのQMSに対して加筆修正が発生し、また追加で作成しなければならないQMSもあります。
本MDR対応QMSひな形を使用することで、より短時間で品質良くMDRに対応したQMSを構築することが出来ます。”]
[blogcard url=”https://ecompliance.co.jp/SHOP/MD-QMS-168.html” title=”【MDR対応QMSひな形】欧州指定代理人用手順書” content=”CEマーキングを貼付し、欧州に医療機器を輸出している医療機器企業は、MDDに置き換わって2021年5月25日までに欧州医療機器規則(MDR:Medical Device Regulation)に対応する必要があります。製造業者(Manufacturer)は、この移行期間中に技術文書を改訂し、新しい要求事項に対応しなければなりません。
MDRは、従来のMDDと能動埋め込み型医療機器指令 Active Implantable Medical Device Directive (90/385/EEC)を統合し置き換えるものです。
MDDからMDRへの改正内容は多岐に渡り、また上市済の製品の対策も必要となるため、強制化に間に合うように計画的に対策を進める必要があります。
MDRは、日本や米国の法規制を参考にして作成され、世界で最も厳しい規制となりました。
しかしながらMDRは条文が難解で非常にわかりにくいのが難点です。
本邦におけるQMS省令や米国FDAのQSRにはない要求事項も含まれます。
そのため、多くのQMSに対して加筆修正が発生し、また追加で作成しなければならないQMSもあります。
本MDR対応QMSひな形を使用することで、より短時間で品質良くMDRに対応したQMSを構築することが出来ます。”]
[blogcard url=”https://ecompliance.co.jp/SHOP/MD-QMS-152.html” title=”【FDA CFR 803対応】MDR手順書” content=”最近、MDR(有害事象報告)に関する指摘が多く出されるようになってきました。 FDAへの有害事象(事故)報告は、所定の期間内に提出しなければなりません。”] [blogcard url=https://xn--2lwu4a.jp/qms-md/ title=”QMS(手順書)ひな形 医療機器関連” ]
]]>
{kind=link}
Comment