

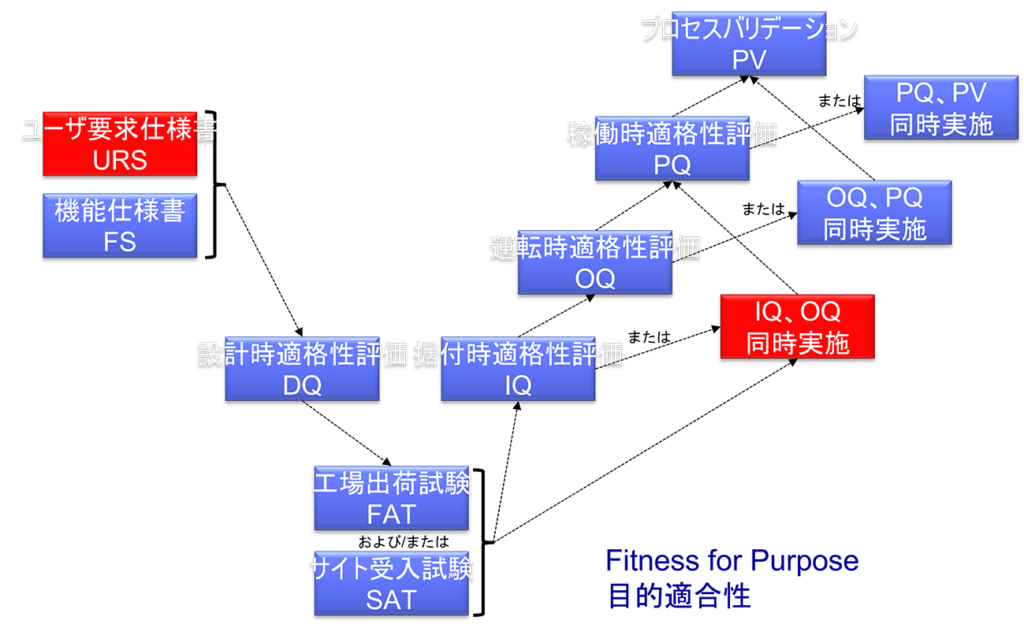
最近ではIQとOQを同時に実施するケースが増えている。
これは、2015年にPIC/S GMP Annex 15 「適格性評価とバリデーション」が改定されてから、IQとOQは同時に実施することが可能となったためである。
OQは、通常IQの後に実施されるが、装置の複雑さによっては、同時にIOQとして実施しても構わない。
またIQとOQの文書を一つにまとめても構わない。
IQは、装置、施設、ユーティリティもしくはシステムについて実施すること。
IQでは下記の項目などを実施する。
1. エンジニアの図や詳述に対して、構成要素、機器類、装置、配管工事やサービスの正しい設置の相互確認
2. あらかじめ定めた条件に対して正しく据付けられたことの検証
3. サプライヤからのオペレーション・作業要領、メンテナンス要件の収集と照合
4. 計測器の校正
5. 建設材料の検証
最近ではIQとOQを同時に実施するケースが増えている。これは、2015年にPIC/S GMP Annex 15 「適格性評価とバリデーション」が改定されてから、IQとOQは同時に実施することが可能となったためである。OQは、通常IQの後に実施されるが、装置の複雑さによっては、同時にIOQとして実施し…
OQは、通常IQの後に実施されるが、装置の複雑さによっては、同時にIOQとして実施しても構わない。
OQでは下記の項目などを実施する。
1. システムが設計どおり操作しているかを確かめるためのプロセス、システム、設備に関する知識を基に開発したテスト
2. 運転の上限及び下限/「ワーストケース」を確認するためのテスト
成功裏にOQが終了すれば、標準作業手順および洗浄手順、作業者訓練や予防的メンテナンスの要件等を最終化できる。
DQ、PQの省略について
最近では構造設備とはカタログ品(既製品)を購入することが多く DQはほぼ実施されなくなった。
またPIC/S GMP Annex 15によると、PQはOQもしくはプロセスバリデーションと合せて実行してもよい。
従って構造設備のバリデーションにおいてDQやPQはほぼ実施されない。
IQとOQが完了してから、PQを実施すること。
しかし、場合によっては、PQはOQまたはプロセスバリデーションと合せて実行してもよい。
PQでは下記の項目などを実施する。
1. ワーストケースのバッチサイズで、製造用原料、適格性評価された代用品、または通常の運転条件で同じ挙動をとることが証明された模擬製品を用いたテスト。
プロセスの管理状態を確認するために用いるサンプリングの頻度が適切であるという根拠を示すこと。
2. 開発段階からの運転範囲を裏付ける文書化された証拠がない限り、テストは意図したプロセスの運転範囲をカバーすること。
FAT/SAT
特に新規であったり複雑な技術が組み込まれた装置は、製薬企業への移送前(IOQ実施前)にベンダー側で評価してもよい。
また装置を設置する前に、ベンダー側でURS/機能仕様書への適合性を確認すること。
適切な理由がある場合、移送や据付作業によって機能に影響がないことが示されれば、文書類のレビュやテストの一部をFATもしくは他の段階で実施することもでき、IQ/OQをオンサイト(製薬企業)で繰り返す必要はない。
つまりベンダーがFATを適切に実施していれば、IOQの実施は(かなり)省略できることになるのである。
なお、FATは製造現場(製薬企業)での装置受入後にSATを実施することによって補完することもできる。
筆者はしばしば製薬企業やベンチャー企業のIOQの実施を支援する。
質問の多くはFAT/SATとIOQとの違いや、PQとプロセスバリデーションの違いである。
かつては適格性評価は製薬企業側において実施しなければならないとされてきた。
多くの場合、製薬企業はベンダーに依頼しそれら文書の作成を実施してきたが、あたかも製薬企業が実施したかのような文書としていた。
しかしながらIOQといった適格性評価を誰が実施するかが重要ではなく、最終的に当該構造設備を使用して製造する製品の品質が保証されることが重要なのである。
多くの場合、IOQの実施はベンダーに依頼することになる。
関連商品
[blogcard url=”https://ecompliance.co.jp/SHOP/O070.html” title=”【VOD】【180分で要点を学ぶ】改正GMPセミナーシリーズ バリデーション編” content=”2021年8月1日からGMP省令が改正されます。改正GMP省令は、ICHやPIC/S等の国際標準のGMP基準に整合されました。
改正GMP省令では、従前のバリデーション基準が廃止され、バリデーション指針となりました。
本邦においては、バリデーション指針またはPIC/S GMP Annex 15「適格性評価とバリデーション」の遵守が求められます。
PIC/S GMP Annex 15 「適格性評価とバリデーション」は、2015年10月1日から改定版が施行されました。
改定版Annex15では、バリデーションに関して、大きな変更が実施されました。
改定版では、2013年1月1日から改定されたAnnex11(コンピュータ化システム)、Annex13(治験薬)との整合性や、ICH-Q8、Q9、Q10との整合性を考慮されました。
プロセスバリデーションについては、2011年のFDAのガイドラインの改定を皮切りに、大きな変革がありました。
製薬企業におけるコンプライアンスコストの上昇は、最終的に患者負担になるため、バリデーション実施に対する負荷を軽減する必要がありました。
そこでベリフィケーションという概念が導入されています。
バリデーションとベリフィケーションはいったい何が違うのでしょうか。またそれぞれの特徴やメリット、デメリットは何でしょうか。
またクオリフィケーション(適格性評価)とプロセスバリデーションの関係はどのようなものでしょうか。
さらにコンピュータ化システムバリデーション(CSV)とクオリフィケーションとプロセスバリデーションの関係はどのようなものでしょうか。
Annex15に記載されている用語はその理解が難しく、新しい用語も増えています。
用語の定義を正しく理解することは、極めて重要です。
現在の製薬工場における施設・設備・装置・機器・ユーティリティのほとんどはコンピュータ化(自動化)されています。
PIC/S GMP Annex11 コンピュータ化システムは、2013年1月1に改定版が施行されました。
コンピュータ化システムにおいての要件にも留意しなければなりません。
製薬工場(GMP)におけるCSV実施の大半は、構造設備や分析機器です。
しかしながら、これまで構造設備や分析機器のCSVについて具体的に解説したセミナーや書籍は皆無でした。
また多くの場合、IQ、OQは製薬企業ではなく、当該ベンダーが実施するというのが一般的になっています。
いったい、どういう風にCSVを実施すべきでしょうか。
構造設備や分析機器の特徴は、ハードウェアが中心で、比較的小さなプログラム(PLCやファームウェア)でコントロールされています。
また、ITアプリケーションと違って、その品質が直感的にわかります。
ITアプリケーションは、複雑かつ大規模なソフトウェアが中心であり、膨大なデータによるテスト(システムテスト、インテグレーションテスト等)を中心に実施します。
一方で、構造設備や分析機器は、DQ、IQ、OQ、PQといったQualification(適格性評価)が中心です。
これまで製薬業界では、プロセスバリデーションで使用されてきたDQ、IQ、OQ、PQといった用語を流用して、CSVを実施してきました。
ではCSVのPQとプロセスバリデーションは何が違うのでしょうか。
そういった、よく寄せられる疑問に答える形でセミナーを進めたいと考えています。 本セミナーでは。難解なバリデーションについて、わかりやすく解説いたします。”]
[blogcard url=”https://ecompliance.co.jp/SHOP/EL-087.html” title=”【セミナービデオ】【180分で要点を学ぶ】改正GMPセミナーシリーズ バリデーション編” content=”2021年8月1日からGMP省令が改正されます。改正GMP省令は、ICHやPIC/S等の国際標準のGMP基準に整合されました。
改正GMP省令では、従前のバリデーション基準が廃止され、バリデーション指針となりました。
本邦においては、バリデーション指針またはPIC/S GMP Annex 15「適格性評価とバリデーション」の遵守が求められます。
PIC/S GMP Annex 15 「適格性評価とバリデーション」は、2015年10月1日から改定版が施行されました。
改定版Annex15では、バリデーションに関して、大きな変更が実施されました。
改定版では、2013年1月1日から改定されたAnnex11(コンピュータ化システム)、Annex13(治験薬)との整合性や、ICH-Q8、Q9、Q10との整合性を考慮されました。
プロセスバリデーションについては、2011年のFDAのガイドラインの改定を皮切りに、大きな変革がありました。
製薬企業におけるコンプライアンスコストの上昇は、最終的に患者負担になるため、バリデーション実施に対する負荷を軽減する必要がありました。
そこでベリフィケーションという概念が導入されています。
バリデーションとベリフィケーションはいったい何が違うのでしょうか。またそれぞれの特徴やメリット、デメリットは何でしょうか。
またクオリフィケーション(適格性評価)とプロセスバリデーションの関係はどのようなものでしょうか。
さらにコンピュータ化システムバリデーション(CSV)とクオリフィケーションとプロセスバリデーションの関係はどのようなものでしょうか。
Annex15に記載されている用語はその理解が難しく、新しい用語も増えています。
用語の定義を正しく理解することは、極めて重要です。
現在の製薬工場における施設・設備・装置・機器・ユーティリティのほとんどはコンピュータ化(自動化)されています。
PIC/S GMP Annex11 コンピュータ化システムは、2013年1月1に改定版が施行されました。
コンピュータ化システムにおいての要件にも留意しなければなりません。
製薬工場(GMP)におけるCSV実施の大半は、構造設備や分析機器です。
しかしながら、これまで構造設備や分析機器のCSVについて具体的に解説したセミナーや書籍は皆無でした。
また多くの場合、IQ、OQは製薬企業ではなく、当該ベンダーが実施するというのが一般的になっています。
いったい、どういう風にCSVを実施すべきでしょうか。
構造設備や分析機器の特徴は、ハードウェアが中心で、比較的小さなプログラム(PLCやファームウェア)でコントロールされています。
また、ITアプリケーションと違って、その品質が直感的にわかります。
ITアプリケーションは、複雑かつ大規模なソフトウェアが中心であり、膨大なデータによるテスト(システムテスト、インテグレーションテスト等)を中心に実施します。
一方で、構造設備や分析機器は、DQ、IQ、OQ、PQといったQualification(適格性評価)が中心です。
これまで製薬業界では、プロセスバリデーションで使用されてきたDQ、IQ、OQ、PQといった用語を流用して、CSVを実施してきました。
ではCSVのPQとプロセスバリデーションは何が違うのでしょうか。
そういった、よく寄せられる疑問に答える形でセミナーを進めたいと考えています。 本セミナーでは。難解なバリデーションについて、わかりやすく解説いたします。”]
[blogcard url=”https://ecompliance.co.jp/SHOP/O071.html” title=”【VOD】(全5コース)【180分で要点を学ぶ】改正GMPセミナーシリーズ【一括受講コース】” content=””] [blogcard url=”https://ecompliance.co.jp/SHOP/EL-088.html” title=”【セミナービデオ】(全5コース)【180分で要点を学ぶ】改正GMPセミナーシリーズ【一括受講コース】” content=””] [blogcard url=”https://ecompliance.co.jp/SHOP/O064.html” title=”【VOD】日本一わかりやすい【超入門】改正GMP省令セミナー” content=”厚生労働省は「医薬品および医薬部外品の製造管理及び品質管理の基準に関する省令」(GMP省令)の一部を改正する省令(厚生労働省令第90号)を発出しました。2021年8月1日の施行を予定しています。
GMPの改正に伴い、この際基礎からGMPを学びたいという方々のために『超入門改正GMP省令セミナー』を企画いたしました。
改正GMP省令では、医薬品品質システム(ICH Q10)、品質リスクマネジメント(ICH Q9)、データインテグリティ、マネジメントレビュ、品質マニュアルなどの難解な用語や概念が出てきます。
これらを日本一わかりやすく解説いたします。
1回聞いただけでは理解できない方にも、アーカイブ配信を繰り返し視聴して頂けたら、きっとGMPを熟知できるようになるでしょう。
本セミナーは、これからGMPを勉強する方にもわかりやすく、基本からお話しを致します。また既にGMPを理解されている方にも再確認して頂ける内容となっています。
本セミナーでは、品質マニュアルのサンプルも配布いたします。”]
2021年8月1日の施行を予定しています。
GMPの改正に伴い、この際基礎からGMPを学びたいという方々のために『超入門改正GMP省令セミナー』を企画いたしました。
改正GMP省令では、医薬品品質システム(ICH Q10)、品質リスクマネジメント(ICH Q9)、データインテグリティ、マネジメントレビュ、品質マニュアルなどの難解な用語や概念が出てきます。
これらを日本一わかりやすく解説いたします。
1回聞いただけでは理解できない方にも、アーカイブ配信を繰り返し視聴して頂けたら、きっとGMPを熟知できるようになるでしょう。
本セミナーは、これからGMPを勉強する方にもわかりやすく、基本からお話しを致します。また既にGMPを理解されている方にも再確認して頂ける内容となっています。
本セミナーでは、品質マニュアルのサンプルも配布いたします。”]
2021年8月1日の施行を予定しています。
GMP施行通知の施行(2013年8月30日)から8年近く経過し、いよいよGMP省令が改正されます。
改正GMP省令は、ICHやPIC/S等の国際標準のGMP基準に整合されました。
特にICH-Q9(品質リスクマネジメント)やICH-Q10(医薬品品質システム)の遵守が求められます。
それにより、品質保証体制の充実が求められることとなりました。
しかしながら、改正GMP省令はPIC/S GMPとの差異も存在します。
いったい何が変わり、どういう要求事項になっているのでしょうか。
改正GMP省令は、PIC/S GMPガイドライン重要項目(6項目)に加え、おおよそ以下の要件が追加されました。
承認事項の遵守(第3条の2)
医薬品品質システム(第3条の3)
品質リスクマネジメント(第3条の4)
交叉汚染の防止(第8条の2)
安定性モニタリング(第11条の2)
製品品質の照査(第11条の3)
原料等の供給者の管理(第11条の4)
外部委託業者の管理(第11条の5)
また、用語の定義が充実しました。
例えば、「医薬品品質システム」、「品質リスクマネジメント」、「安定性モニタリング」、「最終製品」、「参考品」、「保存品」、「是正措置」、「予防措置」、「品質」などが第2条(定義)に追記されます。
いったいどのような手順書(SOP)を作成すれば良いのでしょうか。
【医薬品品質システム】
ICH Q10(医薬品品質システム)の取り込みはグローバルな流れでもあります。
したがって、改正GMP省令においては、ICH Q10の浸透が強く要求されます。
では、医薬品品質システムとはいったい何でしょうか。
医薬品品質システムにおいては、経営層(トップマネジメント)の関与が求められます。
トップマネジメントは、医薬品品質システムの確立と実施の責任を持ちます。
また、定期的にマネジメントレビュによって品質をレビュし、医薬品品質システムの見直しを実施しなければなりません。
それにより、医薬品のライフサイクル全期間での継続的改善を促進することとなります。
また、製造所においては、従来の品質部門に品質保証に係る業務を担う組織(QA)の設置が規定されます。
製造管理者の管理監督の下、品質保証に係わる業務を実際に遂行する組織としての手順書の作成と実施が求められます。
また、外部試験検査機関等の供給者管理も厳格化されます。
供給者監査の実施や供給者における変更管理も把握する必要があります。
さらに品質保証部門(QA)は、是正措置や予防措置(CAPA)を通じて、品質の改善を実施しなければなりません。
【品質リスクマネジメント】
これまでICH-Q9 「品質リスクマネジメントに関するガイドライン」は課長通知として発出されていました。
しかし、改正GMP省令においては、適切に品質リスクマネジメントが活用されるよう、ICHQ9の原則に則して手順書の作成と実施が求められます。
さらに品質リスクマネジメントの適用範囲として、「製品の製造管理及び品質管理」 だけでなく、「製造所における医薬品品質システム(PQS)」も対象となります。
本セミナーは、改正GMP省令と現行のGMP省令の対比表、品質マニュアルのサンプルなど充実した資料を配布し、分かりやすく90分間で改正GMP省令のポイントを解説いたします。”]
[blogcard url=”https://ecompliance.co.jp/SHOP/EL-077.html” title=”【セミナービデオ】改正GMP省令要点セミナー” content=”厚生労働省は「医薬品及び医薬部外品の製造管理及び品質管理の基準に関する省令」(GMP省令)の一部を改正する省令(厚生労働省令第90号 )を発出しました。2021年8月1日の施行を予定しています。
GMP施行通知の施行(2013年8月30日)から8年近く経過し、いよいよGMP省令が改正されます。
改正GMP省令は、ICHやPIC/S等の国際標準のGMP基準に整合されました。
特にICH-Q9(品質リスクマネジメント)やICH-Q10(医薬品品質システム)の遵守が求められます。
それにより、品質保証体制の充実が求められることとなりました。
しかしながら、改正GMP省令はPIC/S GMPとの差異も存在します。
いったい何が変わり、どういう要求事項になっているのでしょうか。
改正GMP省令は、PIC/S GMPガイドライン重要項目(6項目)に加え、おおよそ以下の要件が追加されました。
承認事項の遵守(第3条の2)
医薬品品質システム(第3条の3)
品質リスクマネジメント(第3条の4)
交叉汚染の防止(第8条の2)
安定性モニタリング(第11条の2)
製品品質の照査(第11条の3)
原料等の供給者の管理(第11条の4)
外部委託業者の管理(第11条の5)
また、用語の定義が充実しました。
例えば、「医薬品品質システム」、「品質リスクマネジメント」、「安定性モニタリング」、「最終製品」、「参考品」、「保存品」、「是正措置」、「予防措置」、「品質」などが第2条(定義)に追記されます。
いったいどのような手順書(SOP)を作成すれば良いのでしょうか。
【医薬品品質システム】
ICH Q10(医薬品品質システム)の取り込みはグローバルな流れでもあります。
したがって、改正GMP省令においては、ICH Q10の浸透が強く要求されます。
では、医薬品品質システムとはいったい何でしょうか。
医薬品品質システムにおいては、経営層(トップマネジメント)の関与が求められます。
トップマネジメントは、医薬品品質システムの確立と実施の責任を持ちます。
また、定期的にマネジメントレビュによって品質をレビュし、医薬品品質システムの見直しを実施しなければなりません。
それにより、医薬品のライフサイクル全期間での継続的改善を促進することとなります。
また、製造所においては、従来の品質部門に品質保証に係る業務を担う組織(QA)の設置が規定されます。
製造管理者の管理監督の下、品質保証に係わる業務を実際に遂行する組織としての手順書の作成と実施が求められます。
また、外部試験検査機関等の供給者管理も厳格化されます。
供給者監査の実施や供給者における変更管理も把握する必要があります。
さらに品質保証部門(QA)は、是正措置や予防措置(CAPA)を通じて、品質の改善を実施しなければなりません。
【品質リスクマネジメント】
これまでICH-Q9 「品質リスクマネジメントに関するガイドライン」は課長通知として発出されていました。
しかし、改正GMP省令においては、適切に品質リスクマネジメントが活用されるよう、ICHQ9の原則に則して手順書の作成と実施が求められます。
さらに品質リスクマネジメントの適用範囲として、「製品の製造管理及び品質管理」 だけでなく、「製造所における医薬品品質システム(PQS)」も対象となります。
本セミナーは、改正GMP省令と現行のGMP省令の対比表、品質マニュアルのサンプルなど充実した資料を配布し、分かりやすく90分間で改正GMP省令のポイントを解説いたします。”]
[blogcard url=”https://ecompliance.co.jp/SHOP/O053.html” title=”【VOD】GMP省令改正における「原料等の供給者管理」の対応” content=”PIC/S GMPとの6つのギャップの一つとして、今まで、施行通知で求められていたが、GMP省令が改正され、盛り込まれることになった。今後、原料等の供給者管理は、査察時の重要なポイントとして必ず確認される。しかし、原薬から原料、資材と幅広く、その品質への影響は差があり、取決めや監査について悩まれる点でもある。リスクマネジメントの概念を取り入れ、その取決めや監査のポイントを解説し、より効果的な供給者管理を実施のための手順を探る。”] [blogcard url=”https://ecompliance.co.jp/SHOP/EL-070.html” title=”【セミナービデオ】GMP省令改正における「原料等の供給者管理」の対応” content=”PIC/S GMPとの6つのギャップの一つとして、今まで、施行通知で求められていたが、GMP省令が改正され、盛り込まれることになった。今後、原料等の供給者管理は、査察時の重要なポイントとして必ず確認される。しかし、原薬から原料、資材と幅広く、その品質への影響は差があり、取決めや監査について悩まれる点でもある。リスクマネジメントの概念を取り入れ、その取決めや監査のポイントを解説し、より効果的な供給者管理を実施のための手順を探る。”] [blogcard url=https://xn--2lwu4a.jp/qms-alex/ title=”QMS(手順書)ひな形 医薬品関連” ]]]>
{kind=link}
Excellent goods from you, man. I have have in mind your stuff previous to and you’re just too great.
I really like what you’ve bought right here, really like what you
are stating and the way in which during which you are saying it.
You make it entertaining and you continue to care for to keep it
smart. I cant wait to read far more from
you. This is actually a terrific website.