医療機器というのは、人の生命に直接的な影響を与える。
医療機器として認められるためには、品質(Quality)と有効性(Efficacy)と安全性(Safety)の三つ、SEQの証明(確保)が必要である。
SEQを証明した申請資料を当局または認証機関に提出して、認められなければならない。
医療機器というのは、人の生命に直接的な影響を与える。医療機器として認められるためには、品質(Quality)と有効性(Efficacy)と安全性(Safety)の三つ、SEQの証明(確保)が必要である。SEQを証明した申請資料を当局または認証機関に提出して、認められなければならない。…
医療機器の規制目的(日本の厚生労働省、米国FDA、欧州とも目的は同じ)
「良質高度な医療製品の国民への提供」(推進)と「医療機器の安全性と効果効能を規制することで国民の健康被害を防止」(規制)の2つの目的があり、自動車のアクセルとブレーキに例えることができる。
薬事法から医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律への改定
薬事法は、昭和35年に制定された。
薬事という名前により医薬品を想像させるため、「薬事法等の一部を改定する法律」(平成25年11月27日、平成25年法律第84号。「改正法」)により、「医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律」(以下、薬機法)に改められた。
特徴的なことは、2つあり、タイトルに医療機器という名前が入ったことと、品質、安全性、有効性の3つの要素が盛り込まれたことである。
「医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律」(以下、薬機法)
薬機法で規制される製品は、医薬品、医薬部外品、化粧品、医療機器及び再生医療等製品の5つである。
薬機法には2つの目的があり、一つは、品質、有効性及び安全性の確保並びにこれらの使用による保健衛生上の危害の発生及び拡大の防止のために必要な規制を行う(ブレーキ)。もう一つは、医療上特にその必要性が高い医薬品医療機器及び再生医療等製品の研究開発の促進(アクセル)である。
厚生労働省又は経済産業省が製品の研究開発を促進するアクセルの役割を担い、PMDA(独立行政法人医薬品医療機器総合機構)が審査・承認や必要な規制をするブレーキの役割を担っている。
医療機器製造販売業の許可、品質保証体制、安全管理体制の構築
薬機法には、「次の各号のいずれかに該当するときは、前条第一項の許可を与えないことができる。」と記載されており、これは厚生労働省令で定める基準に適合しなければならないという意味である。
基準の一つ目は、「申請に係る医療機器又は対外診断用医薬品の製造管理又は品質管理に係る業務を行う体制が、厚生労働省令で定める基準に適合しないとき。」と記載されており、これは厚生労働省令第94号(体制省令)により、体制を作らなければならないということである。
基準の二つ目は、「申請に係る医療機器又は対外診断用医薬品の製造販売後安全管理の方法が、厚生労働省令で定める基準に適合しないとき。」との記載があり、厚生労働省令第135号GVP(Good Vigilance Practice)省令に適合しなければならない。
国は、医療機器又は体外診断用医薬品の製造管理又は品質管理に係る業務を行う体制の基準(体制)、製造管理及び品質管理の基準(QMS)、医療機器の市販後安全管理に関する基準(GVP)を、省令として公布している。
医療機器の製造販売業許可業者は、これらの省令にしたがって、適切に、医療機器の品質管理・安全管理を行わねばならない。
その体制が整っていることが、「許可の取得」「許可の維持」の要件となっている。
規制する3つの要素
医療機器を規制する要素は3つある
- 市販適合性評価(承認、認証、届出等)
市販適合性評価には、国(PMDA)が承認するものと、民間の認証期間(ノーティファイドボディ(NB))が認証するもの、届出だけで流通させられるものがある。
承認と認証の申請手順は全く同じである。
医療機器はそのリスクに応じてクラス分類がなされており、クラスI、クラスII、クラスIII、日本の場合はクラスⅣまである。
クラスIは、例えば、絆創膏のように、届出をすることによってすぐ市場に出すことができる。
クラスIIの医療機器に関しては、多くの製品が民間の認証機関による認証を取得すれば市場に出すことができる。
クラスⅢの機器の多くは、PMDAの承認審査に合格する必要がある。
この市販適合性評価を行うということが一つ目の規制である。
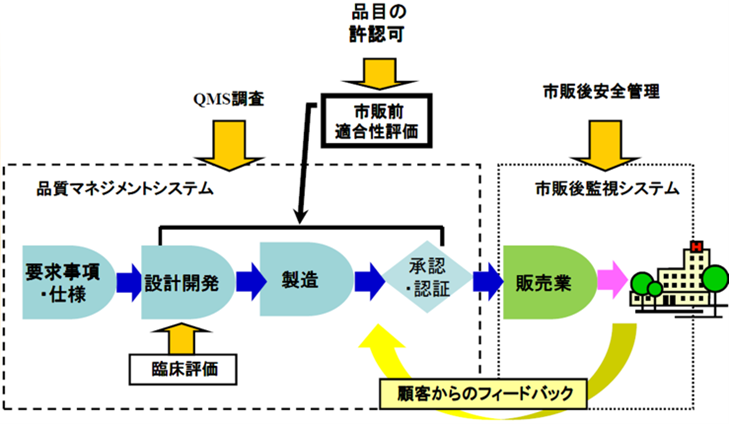
上市する前に、製品(品目)自体が安全性、かつ効果・効能が規定要求事項を満足している必要がある。 - 品質の確保、QMS(品質マネジメントシステム)調査(QMS省令、体制省令)
新規の医療機器を申請または認証審査された場合に、QMR省令に従った当局のQMS調査(査察)がある。
医療機器の製品ライフサイクル全体の管理が適正に実施されていることが調査され
その際に品質が適切に管理され、品質の良い製品が出荷されるようになっていることが調査される。
QMS適合性調査には、製造所に実際に調査官の方が訪問する実地調査と、設計文書や製造文書を当局または認証機関に送って書面で調査するという信頼性調査の2通りがある。
ISO-13485認証を取得していれば、実地調査が軽くなることや免除されることも多々ある。
日本の場合は、特に医療機器製造販売業や医療機器製造業を取るためにISO-13485の認証を必須とはしないが、ISO-13485の認証を受けていれば、QMS適合性調査等では有利になる。 - 市販後安全管理(GPV省令)
上市後の製品の安全情報を収集し、分析し、評価して、必要な措置(回収・不具合)を行う。(GVP省令に基づき実施する)
例えば回収や不具合報告で通知書を撒くことや、厚生労働省に報告する又はPMDAに報告する等の措置を取らなければならない。
品質システム規格の歴史とQMS省令
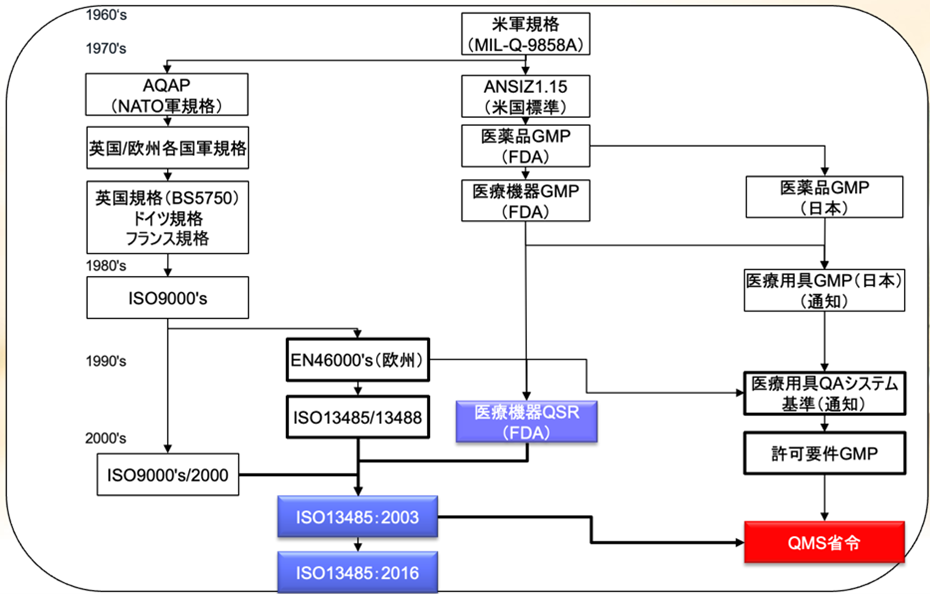
医療機器に関する規制要件は、日米欧でそれぞれ作られてきた。
米国では、ケミカル由来の医薬品とデバイス由来の医療機器では規制の内容が違うため、医薬品のGMPから医療機器のGMPが別れ、その後1997年に米国FDAは医療機器QSR(Quality System Regulation:品質システム規則)という新しい規制要件を発出した。
欧州ではISO9000から医療機器に特化して作られた国際規格ISO-13485が独立した。
ISOの加盟各国によって批准されたISO-13485のバージョン1.0が2003年版である。
日本では、医療用具GMPがあり、医療機器の許可要件GMPになり、その後QMS省令というものに名前が変わったが、2015年度の薬事法の一部改正でISO-13485の2003年版と整合をとることになった。
今般、ISO-13485の2016年版(ISO-13485のバージョン2.0)が発出されが、これは1997年に米国FDAが発行したQSRとほぼイコールである。
日本でも5年遅れて2021年3月26日にQMS省令とISO-13485の2016年版との整合を図った改正QMS省令が公布され、経過措置期間は 3年間で、2024年3月26日(改正省令の施行の日から起算して3年を経過する日)には新QMS省令を遵守しなければならない。
このISO-13485の2016年版に準拠することにより、日米欧の要求事項に適合することになる。 ただし、これは品質面だけのことであり、安全性・有効性の問題は別の規格がある。
お役立ち動画
医療機器の設計・開発・申請における規制要件入門
~品質、有効性及び安全性の確保~
▶ 1講 医療機器と規制要件
▶ 2講 医療機器の種類
▶ 3講 医療機器と品質
▶ 4講 医療機器と安全性
▶ 5講 医療機器と有効性
▶ 6講 医療機器申請と当局査察
関連商品
[blogcard url=”https://ecompliance.co.jp/SHOP/QMS-LIVE.html” title=”(全16講)医療機器QMS規制入門セミナー【一括受講コース】” content=”医療機器企業に初めて入社・転職した人向けの規制要件入門コースです。QMSを中心に解説を行います。
本邦では、医療機器企業(製造販売業)においては、QMS省令(体制省令)およびGVP省令に則った体制の構築が出来ていなければなりません。
またQMS省令に従って、QMS(Quality Management System:品質管理システム)が文書化されており、適切にQMSに従って運用されており、それら活動の記録が作成されていなければなりません。
QMS省令は、ISO 13485をもとに作成されています。
ISO 13485は、マネジメント(経営者)の責任・リソース(資源)の配分(Plan)、製品実現(Do)、監視測定(Check)、改善(Action)といったPDCAサイクルで構成されています。
PDCAサイクルがあるということは、品質保証システムがあるということで、「今日」よりも「明日」、「明日」よりも「明後日」の方が品質が向上していくという証明となります。
規制当局は「継続的な改善」を求めています。
日進月歩の医療機器においては、常に設計変更が求められ、品質の改善も求められています。
本講座は16講(2時間/講)にわたって、医療機器業界にけるQMSに関する規制要件の内容を初心者にとって分かりやすく、丁寧に解説いたします。”]
[blogcard url=”https://ecompliance.co.jp/SHOP/QMS-MHLW-00.html” title=”【2021年度改正QMS省令対応】QMSひな形一式” content=”厚生労働省は、2021年3月26日付で「医療機器及び体外診断用医薬品の製造管理及び品質管理の基準に関する省令の一部を改正する省令」(令和3年厚生労働省令第60号)を公布しました。
経過措置期間は 3年間であり、2024年3月26日(改正省令の施行の日から起算して3年を経過する日)には新QMS省令を遵守しなければなりません。
今回の改正の趣旨は、QMS省令と医療機器の品質マネジメントシステムの国際規格であるISO13485:2016と整合を図ることです。
改正前のQMS省令はISO13485:2003年版と整合させており、最新の国際規格からは遅れていました。
厚生労働省は、2021年3月26日付で新QMS省令に関する逐条解説も公表しています。 「医療機器及び体外診断用医薬品の製造管理及び品質管理の基準に関する省令の一部改正について」(薬生発0326第10 号)
イーコンプライアンスでは、下記のお役立ち資料を作成し配布しております。
・改正QMS省令(本文)
・改正QMS省令と現行のQMS省令の対比表
・改正QMS省令逐条解説
・品質マニュアル(QMS省令2021年版対応)
・改正QMS省令手順化要求差異配布
・製品標準書新旧対応表配布
ご希望の方はこちらからダウンロードをお願いいたします。
すでに現行QMS省令に準拠したQMSを構築されている方で、改正対応が必要な方
これから医療機器に参入する方
にオススメのひな形セットです。”]
[blogcard url=https://xn--2lwu4a.jp/qms-md/ title=”QMS(手順書)ひな形 医療機器関連” ]
]]>
{kind=link}
This is my first time visit at here and i am really impressed
to read everthing at one place.