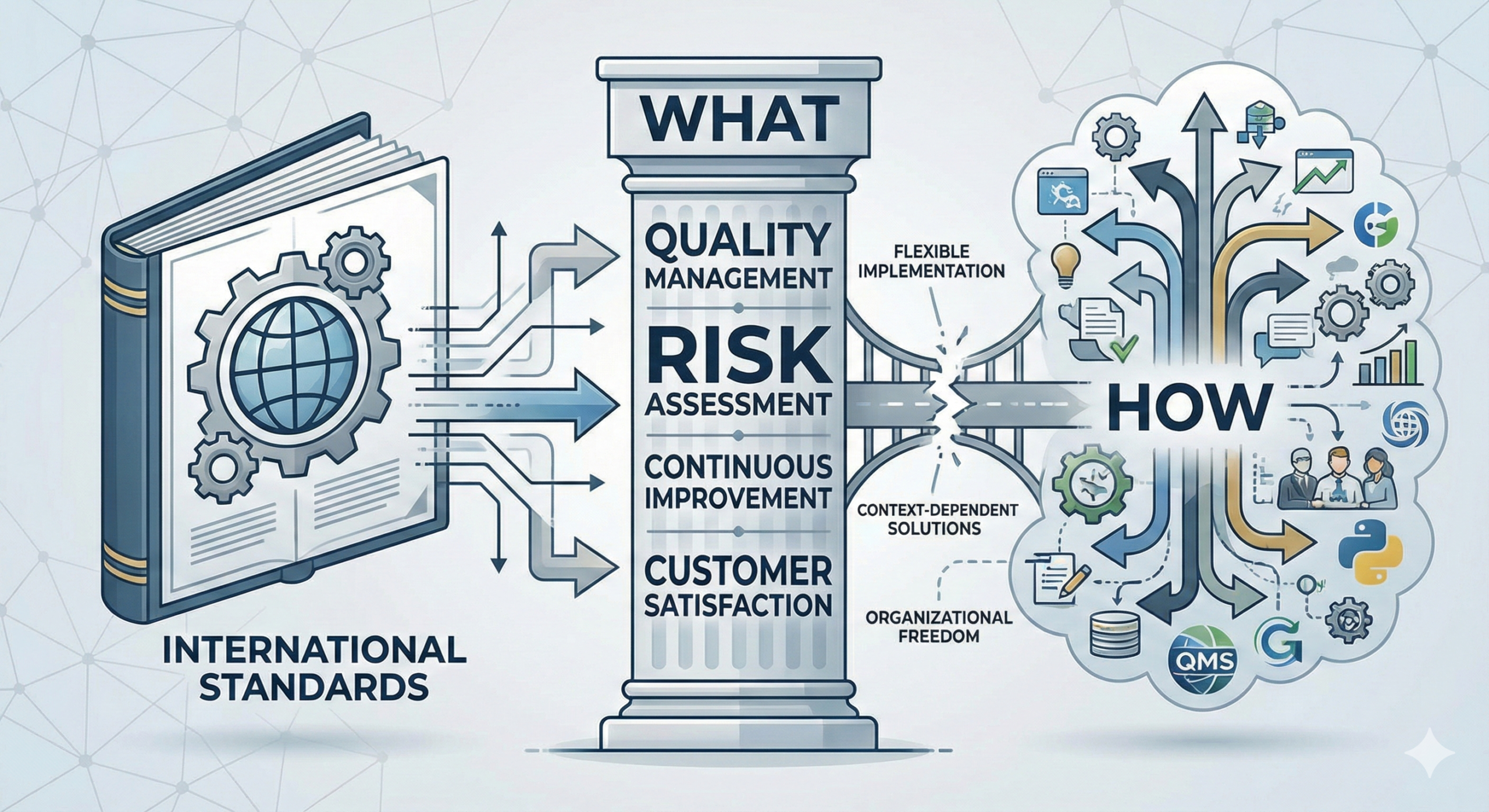
Why International Standards Don’t Explicitly Specify “How”

Introduction
For companies worldwide to guarantee the quality of products and services and operate business safely, compliance with international standards is essential. However, when encountering international standards for the first time, many people wonder: “Why aren’t specific procedures and methods (How) written into these standards?”
For companies worldwide to guarantee the quality of products and services and operate business safely, compliance with international standards is esse…
This question stems from a fundamental principle of international standardization. International standards focus on “what needs to be accomplished” (What) while implementation methods (How) are presented as examples rather than prescriptions. In this article, we explore the reasoning behind this design, practical applications, and the role of Technical Reports and Technical Specifications as implementation guidance—explained clearly for beginners while maintaining professional rigor.
Why International Standards Emphasize “What” Over “How”
The Fundamental Design Principle of Standards
International standards deliberately avoid prescribing specific means or implementation methods, instead enumerating only the requirements (specifications) that must be achieved. This is an intentional design choice grounded in multiple strategic considerations.
1. Adaptability to Technological Evolution
Technology evolves rapidly. Consider IEC 62304 (Medical device software lifecycle processes), which does not prescribe specific programming languages, tools, or development methodologies. Why? Because after a standard’s publication, new languages and frameworks emerge—and generative AI-driven development approaches proliferate. If a standard rigidly defined “which specific methods must be used,” it would become obsolete, unable to accommodate technological advances.
By limiting itself to “What,” the standard maintains the essential requirement of “establishing a safe and effective software development process with activities executed at each lifecycle stage,” while allowing organizations to freely select their implementation approaches. This ensures the standard remains relevant across technology generations.
2. Adaptability to Different Industries, Sectors, and Organizational Characteristics
The same ISO 9001 applies to vastly different companies: medical device manufacturers, automotive suppliers, food producers, and software enterprises. Their business processes, regulatory requirements, market environments, and organizational sizes differ dramatically.
For example, ISO 9001 requires “controlling documents,” but does not specify how. A large automotive manufacturer might deploy a comprehensive document management system integrated with enterprise resource planning (ERP) software, while a startup might use cloud-based collaboration tools (Google Drive, Microsoft 365). Both satisfy the “What” (control documents), and both comply with ISO 9001.
FDA medical device regulations follow a similar principle. The FDA Quality System Regulation (QSR) and FDA guidance documents specify what companies must achieve, but decisions about which specific processes or procedures to employ rest with the organization. Whether a medical device company pursues FDA clearance, Japanese PMDA approval, or EU MDR conformity, all can satisfy ISO 13485 requirements through different implementations.
3. Scalability and Feasibility Across Organizations
By specifying “What,” standards become applicable to large enterprises and small businesses alike, to complex industries and simple ones. This universal applicability is one reason international standards achieve such widespread adoption globally.
Bridging the Gap: Technical Reports, Technical Specifications, and Implementation Guidance
While standards appropriately focus on “What,” this approach may create implementation uncertainty: “So what exactly should I do?” To address this question, multiple guidance tools exist.
Technical Reports (TR) and Technical Specifications (TS)
Within the ISO/IEC system, Technical Reports (ISO/IEC TR) and Technical Specifications (ISO/IEC TS) function as complementary guidance to their corresponding base standards.
Characteristics of Technical Reports (TR):
- Explain the background, theoretical rationale, and design philosophy of the standard
- Provide implementation examples and best practices
- Include information addressing emerging technology trends and industry developments
- Serve as reference material with no mandatory force
Characteristics of Technical Specifications (TS):
- Provide more detailed implementation guidance
- Offer more concrete direction on how to implement standard requirements
- Generally carry greater prescriptive weight than TRs
For example, related Technical Reports to the ISO/IEC 80001 series (Network security management for medical devices) elaborate on network architectures, threat modeling, and concrete risk management implementation methods.
For information security management systems (ISMS), numerous Technical Reports (such as ISO/IEC TR 27009) accompany ISO/IEC 27001 (requirements), providing sector-specific implementation guidance for financial institutions, healthcare organizations, cloud service providers, and other domains.
Additional Guidance and Implementation Support
Beyond Technical Reports, several forms of guidance exist:
ISO/IEC 27002 (Code of Practice for Information Security Controls) Supporting ISO/IEC 27001 requirements, this document provides specific security controls and detailed implementation methods. The 2022 edition describes 114 security controls in detail.
Regulatory Authority Guidance (FDA, PMDA, EMA) Regulatory agencies issue guidance explaining how manufacturers should implement regulatory requirements. For example, FDA guidance on “21 CFR Part 11 Electronic Records; Electronic Signatures” provides detailed instruction on how electronic record and signature systems should be designed and operated.
Industry Association Guidance and Implementation Standards Organizations such as AAMI (Association for the Advancement of Medical Instrumentation), IMDRF (International Medical Device Regulators Forum), and EDMA (European Medical Device Association) publish guidance helping member organizations implement regulatory requirements and standards.
Understanding Through Practice: “What” and “How” in Medical Device Standards
Let’s examine this principle more concretely using medical device regulation as an example.
ISO 13485 (Medical device quality management systems) specifies design management requirements as follows:
Standard Requirements (What): “The organization shall plan and manage medical device design and development, including design stages, reviews, verification, and validation.”
However, the standard does not specify:
- The exact number of design development stages
- What deliverables must be created at each stage
- Which tools or systems to use
- What form design reviews should take (on-site, remote, hybrid)
One company might adopt a staged gate-based design process (five gates) using digital collaboration tools for parallel design development. Another might employ a more linear waterfall approach (three stages) emphasizing in-person design reviews. Whether pursuing FDA clearance, PMDA approval, or EU MDR conformity, all can satisfy ISO 13485 requirements.
Similarly with IEC 62304 (Medical device software development). When the standard requires “conducting software unit testing with defined test cases and expected results,” it specifies the “What” but not the implementation details—such as how to integrate automated testing tools in an agile development environment or which systems to connect to test management tools.
Application of Design Philosophy: Risk Management Standards
ISO 14971 (Medical device risk management) applies the same principle. The standard requires “conducting risk assessment and evaluating the effectiveness of risk controls against acceptable risk criteria,” but delegates these implementation choices to the organization:
- Which risk assessment method to use (FMEA, FTA, hazard analysis, etc.)
- How to define and calculate acceptable risk criteria
- What format to use for risk management files (paper, digital, cloud-based)
The standard enables medical device manufacturers to select the methods most appropriate to their product characteristics, foreseeable use cases, and patient populations.
Distinction Between Regulatory Requirements and Standards: Understanding “Shall” and “May”
An important distinction must be made. Requirements in international standards (ISO/IEC) typically use “shall” language, but implementation approaches employ flexible expressions like “may” and “recommended.”
Conversely, regulatory requirements (FDA 21 CFR, EU MDR, PMDA guidance) sometimes include more specific “How” details. For example, FDA 21 CFR Part 11 specifies concrete technical requirements for electronic records and signatures—audit trails, timestamps, encryption. However, not all regulatory requirements prescribe “How” in such detail. Modern risk-based regulations increasingly emphasize “interactive regulatory compliance,” where organizations propose implementation approaches aligned with their circumstances and seek regulatory authority agreement.
Current Trends: AI/ML Medical Devices and Standard Adaptability
The regulatory guidance-setting process for AI/machine learning (ML)-enabled medical devices starkly illustrates the “What” and “How” challenge.
Since 2023, the FDA, PMDA, and EMA have successively released guidance on AI/ML medical devices. Yet much of this guidance cannot specify all implementation methods—technologies evolve too rapidly. Instead, guidance states “What”: “Establish continuous learning systems to detect unexpected performance degradation” and “Assess model bias and robustness,” with implementation methods remaining as examples.
This demonstrates how standards and regulations evolve together, and how the flexibility of foundational standards like ISO 13485 and IEC 62304 proves critical for addressing emerging technologies.
Practical Example: How Technical Reports Are Used
Imagine a medical device company faces the question: “How should we plan and conduct software validation?” The organization might reference guidance in this sequence:
- IEC 62304 Base Standard: Confirm software validation is a mandatory requirement, and validation planning and reporting are necessary.
- Related Technical Reports: Learn validation strategies (black-box testing, white-box testing, integration testing), testing environment setup, and user environment testing implementation.
- FDA, PMDA, and EMA Guidance: Confirm specific regulatory expectations (for example, conditions for conducting validation in clinical settings).
- Company-Specific Technical Specifications and Product Risk: Based on above guidance, develop a validation plan proportionate to product complexity and market risk.
This approach enables companies to satisfy “What” while implementing the “How” best suited to their circumstances.
Conclusion
The emphasis of international standards on “What” rather than explicit “How” is not a design flaw but a strategic choice ensuring standards’ lasting utility, applicability, and capacity to accommodate innovation.
This design philosophy achieves:
- Temporal Durability: Essential requirements remain valid across technology generations, unconstrained by rapid technical change.
- Spatial Applicability: Standards apply across industries, sectors, organizational sizes, and geographic regions.
- Maintained Competitiveness: Organizations can apply creative problem-solving and organizational strengths to achieve value beyond mere standard compliance.
Meanwhile, Technical Reports, industry guidance, regulatory authority guidance, and best practice examples provide ongoing navigation of “How,” enabling both newcomers and experienced professionals to discover practical pathways for implementing standard requirements.
Standards compliance represents a critical element for organizational credibility and quality assurance, but it simultaneously constitutes a strategic choice of frameworks supporting operational improvement and business value creation. Only when organizations correctly understand the relationship between “What” and “How” does the true value of international standards become manifest.
Related Articles
- Classification System of International Standards
- Understanding Technical Reports in International Standards
- The Amended GMP Ordinance and International Standards Harmonization
- Differences in Approach Between JIS Standards and FDA Guidance: Current State of International Harmonization
- Understanding the Concept of “Organization” in ISO Standards
- Reflections on the Revised GMP Ministerial Ordinance: From Standards to Professional Practice
{kind=link}
Comment