What Are the Unique Characteristics of the Risk Model for In Vitro Diagnostic Medical Devices (IVDs)?

When people hear the term “medical device,” many envision surgical instruments, pacemakers, and other devices that directly contact a patient’s body. However, among the medical devices playing crucial roles in healthcare settings, there exist those that significantly impact patient health without any direct physical contact with patients. These are In Vitro Diagnostic medical devices, commonly known as IVDs.
When people hear the term “medical device,” many envision surgical instruments, pacemakers, and other devices that directly contact a patient’s body. …
IVDs encompass the instruments and reagents used in various tests we receive at hospitals, such as blood tests, urine tests, and infectious disease testing. At first glance, these may appear to pose minimal direct risk to patients. However, due to their unique risk model, they occupy a distinctive position within medical device regulation. Understanding this specialized risk structure has become increasingly important as regulatory frameworks worldwide, including the European Union’s In Vitro Diagnostic Regulation (IVDR) and the United States’ Clinical Laboratory Improvement Amendments (CLIA), have evolved to address the complexities inherent in diagnostic testing.
The Essential Nature of IVD’s “Invisible Risks”
Indirect Yet Critical Risks
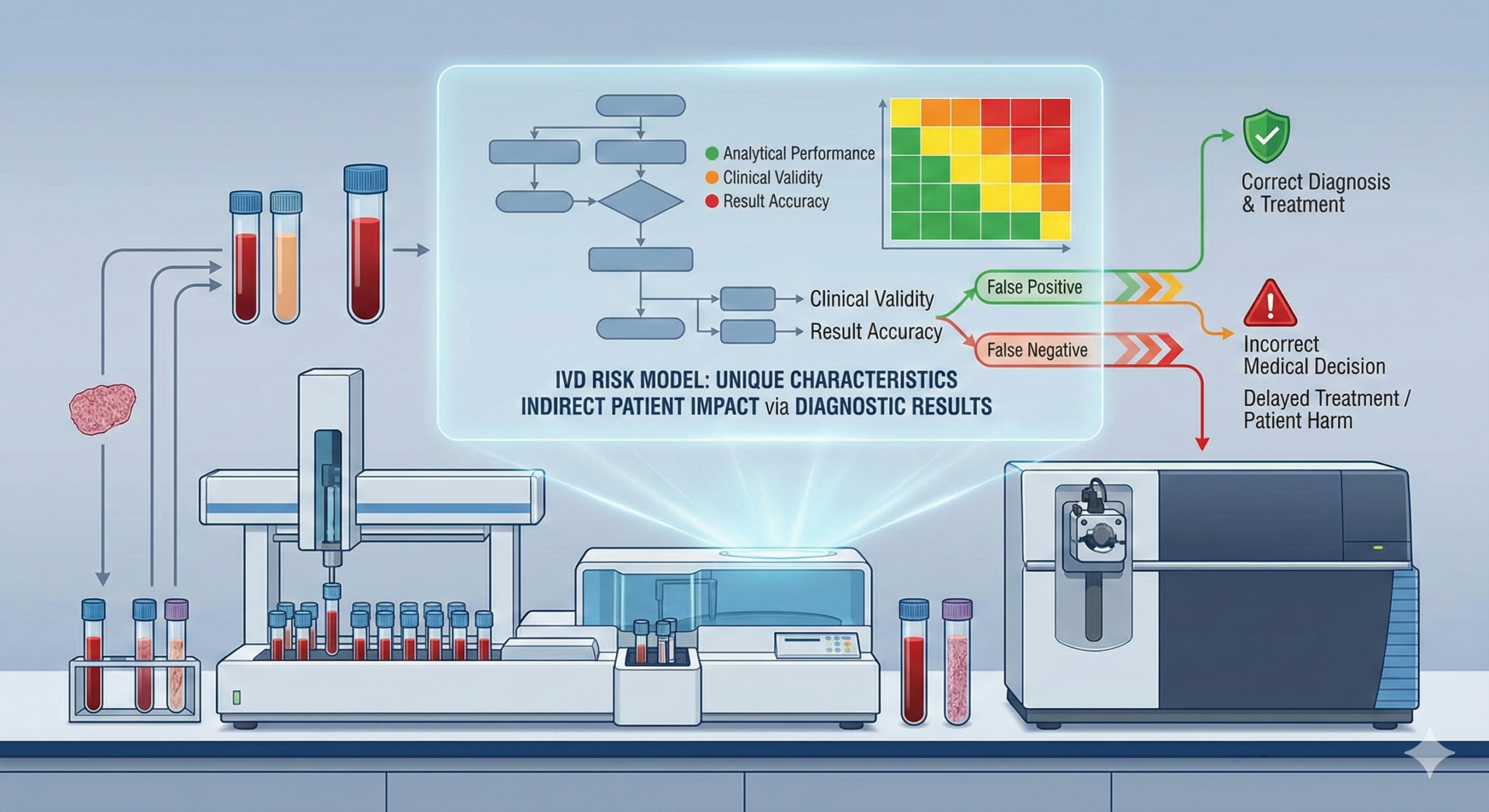
The most distinctive characteristic of IVDs lies in the fact that the devices themselves do not cause physical harm to patients. While there may be pain from a needle stick during blood collection, the testing instruments or reagents themselves do not injure the patient’s body. However, herein lies a significant pitfall.
Consider, for example, a case where an HIV test produces a false-negative result. A patient who is actually infected but receives a “negative” result loses the opportunity to receive appropriate treatment and may unknowingly increase the risk of transmission to others. Similarly, if a false-negative result occurs in cancer tumor marker testing, opportunities for early detection and treatment are lost, potentially having serious implications for the patient’s prognosis.
In this way, health hazards from IVDs occur indirectly through “erroneous test results,” yet their impact can sometimes be more severe than harm from direct-contact medical devices. This fundamental principle underlies the risk-based classification systems adopted by major regulatory authorities worldwide. Under the IVDR, which came into application in May 2022, IVDs are classified into four risk classes (A, B, C, and D) based on the potential consequences of false-positive or false-negative results. Similarly, the U.S. Food and Drug Administration (FDA) classifies IVDs into three classes (I, II, and III) according to risk level, with approximately 50% classified as Class I (lowest risk), 45% as Class II (moderate risk), and 5% as Class III (highest risk requiring premarket approval).
The Cascading Effects of Misdiagnosis
IVD risks affect patients in three primary forms, each with potentially severe consequences for patient safety and public health.
First, misdiagnosis leads to inappropriate treatment. Based on incorrect test results, unnecessary treatments or surgeries may be performed. For instance, a false-positive result could lead a healthy person to undergo chemotherapy. The psychological and physical burden of such inappropriate interventions extends beyond immediate harm, potentially causing long-term complications and diminished quality of life.
Second, treatment delays occur when false-negative results cause necessary treatment to begin late, allowing the disease to progress. This delay can be fatal, particularly in progressive diseases. In oncology, for example, delayed diagnosis due to false-negative tumor markers can mean the difference between early-stage cancer amenable to curative treatment and advanced disease requiring more aggressive, less effective interventions. Similarly, in infectious disease testing, false-negative results for conditions such as tuberculosis or HIV not only harm the individual patient but also pose public health risks through continued transmission.
Third, psychological impacts manifest when incorrect test results cause unnecessary anxiety in patients or, conversely, create a false sense of security. These psychological effects significantly impact patients’ quality of life (QOL) and may influence subsequent health-seeking behaviors. A false-positive result for a serious disease can lead to severe emotional distress, relationship problems, and even unnecessary medical interventions before the error is discovered. Conversely, false-negative results may cause patients to ignore warning symptoms or delay seeking medical attention, potentially with catastrophic consequences.
The severity of these risks varies depending on the intended use of the IVD. For instance, devices used to screen blood donations for transmissible agents such as HIV, hepatitis B, or hepatitis C are classified as Class D under IVDR due to the life-threatening nature of these diseases and the high risk of propagation if undetected. Similarly, devices used in critical decision-making scenarios, such as determining the infectious load for monitoring life-threatening diseases, carry the highest risk classification.
Complexity Created by Four Stakeholders
A Stakeholder Structure Different from Conventional Medical Devices
General medical devices typically involve three primary parties: manufacturers, healthcare professionals, and patients. However, in the case of IVDs, an important fourth player enters the equation—the “laboratory technician” or “clinical laboratory scientist”—creating a four-party structure.
The roles of these four stakeholders can be characterized as follows. Manufacturers assume responsibility for IVD device and reagent development, manufacturing, and quality control. They must ensure that their products meet applicable regulatory requirements, including those specified under IVDR in Europe, FDA regulations in the United States, and CLIA standards for laboratory use. Under the IVDR, manufacturers of Class B, C, and D devices must undergo conformity assessment by Notified Bodies, with only Class A devices permitted self-declaration of conformity.
Laboratory technicians and clinical laboratory scientists represent the specialized professionals who actually perform tests and interpret results. In the United States, these professionals must meet specific qualifications under CLIA regulations, which categorize laboratory testing into waived, moderate complexity, and high complexity levels. Laboratories performing moderate and high complexity testing must employ personnel meeting defined education, training, and experience requirements. These requirements were updated in 2025 as part of CLIA’s first major regulatory revision in decades, with stricter standards for personnel qualifications and competency assessment.
Physicians bear responsibility for determining diagnostic and treatment strategies based on test results. However, they often lack detailed knowledge of the specific IVD products used, their performance characteristics, and their limitations. This creates a critical information gap that can lead to misinterpretation of results.
Patients receive testing and undergo medical treatment based on the results. Unlike traditional medical devices where patients directly experience device function or malfunction, patients receiving IVD testing may remain unaware of issues with test quality or accuracy until the consequences become apparent through clinical outcomes.
This four-party structure complicates information transmission pathways, increasing the possibility of errors at each stage. The complexity is further compounded by the fact that different regulatory authorities may have jurisdiction over different aspects of the IVD ecosystem. For example, in the United States, the FDA regulates IVD devices as medical devices, while the Centers for Medicare & Medicaid Services (CMS) regulate clinical laboratories through CLIA. The Centers for Disease Control and Prevention (CDC) also plays a role in developing technical standards and laboratory practice guidelines.
The Challenge of Information Asymmetry
A particularly problematic issue involves information asymmetry between physicians and testing. Due to the demands of daily clinical practice, many physicians lack time to read in detail the package inserts of IVD products used. Consequently, it is common for diagnoses to be made based solely on the numerical values or determinations of test results.
However, several critical pieces of information exist regarding testing that physicians may not fully consider in their clinical decision-making. Test sensitivity and specificity determine how accurately the test performs—specifically, the test’s ability to correctly identify true positives (sensitivity) and true negatives (specificity). These performance characteristics are fundamental to interpreting test results, yet they vary significantly among different IVD products and testing methodologies.
The measurement range and detection limits define the quantitative boundaries within which the test can provide reliable results. Values outside this range may be reported as “below detection limit” or “above linearity,” requiring careful clinical interpretation. Understanding these limitations is essential for appropriate clinical decision-making, particularly when monitoring disease progression or therapeutic response.
The effects of interfering substances represent another critical consideration. Many factors can interfere with IVD test results, including medications, dietary components, endogenous substances such as bilirubin or lipids, and hemolysis. Package inserts typically list known interfering substances, but physicians may not be aware of these interactions when ordering or interpreting tests.
The impact of specimen storage conditions can significantly affect test results. Some analytes are stable for extended periods, while others degrade rapidly if specimens are not properly handled. Temperature, exposure to light, and time between collection and testing all influence result accuracy. Laboratory professionals understand these requirements, but this information may not reach the ordering physician.
When diagnoses are made without this information being adequately conveyed to physicians, the risk of misinterpreting test results increases substantially. This information gap has motivated recent regulatory changes, including enhanced labeling requirements under IVDR and increased emphasis on communication between laboratories and clinical users in CLIA’s 2025 updates.
Why Information Accuracy Is Lifeline
The Importance of Package Inserts and Instructions for Use
In IVDs, package inserts and Instructions for Use (IFU) are not merely explanatory documents. They represent critical safety mechanisms for correctly interpreting test results and protecting patient safety. These documents must contain comprehensive information to guide appropriate use and result interpretation.
Performance characteristics constitute the foundation of the IFU, including sensitivity, specificity, precision, accuracy, analytical measurement range, and clinical performance data. Under IVDR, these characteristics must be established through performance evaluation studies conducted in accordance with the device’s intended use and supported by clinical evidence. The level of clinical evidence required varies by device class, with Class D devices requiring the most extensive data.
Precautions for use must clearly identify conditions that may lead to false-positive or false-negative results. This includes information on specimen types, collection procedures, storage requirements, and any known interfering substances or cross-reactivity issues. The IVDR requires manufacturers to provide clear information on any limitations of the device and circumstances where the device should not be used.
Limitations of the test must be explicitly stated, including situations where the test may fail to detect analytes or where results require careful interpretation. For example, screening tests with high sensitivity may produce false positives that require confirmatory testing. The IFU must guide users on appropriate interpretation and next steps.
Clinical significance explains the medical meaning of test results in the context of patient care. This includes information on positive and negative predictive values, which depend not only on test performance but also on disease prevalence in the tested population. Understanding these concepts is crucial for appropriate clinical decision-making.
In the United States, the FDA has recently increased regulatory oversight of laboratory-developed tests (LDTs) through a final rule published in 2024. This rule phases out the Agency’s general enforcement discretion policy for LDTs over a four-year period ending in 2028. Beginning May 6, 2025, laboratories offering LDTs must comply with medical device reporting requirements, including adverse event reporting and quality system complaint file requirements. By May 6, 2026, they must comply with all other medical device requirements except quality system design controls and premarket review. This regulatory change reflects recognition that clear, accurate information is essential for patient safety regardless of whether an IVD is commercially manufactured or developed within a laboratory.
Key Points of Risk Management
Risk management for IVDs requires particular attention to several critical areas, each supported by regulatory requirements and quality management principles.
Ensuring traceability represents a fundamental requirement for IVD risk management. When abnormalities appear in test results, systems must exist to track the reagent lot used, instrument calibration status, testing personnel, and all relevant environmental conditions. The IVDR requires full traceability from raw materials through manufacturing and distribution to end use. In the United States, CLIA regulations mandate that laboratories maintain detailed records enabling complete reconstruction of the testing process. These traceability systems enable rapid response when issues are identified, including recall of affected test results, investigation of root causes, and implementation of corrective actions.
Thorough quality control maintains testing reliability through systematic monitoring and assessment. This includes internal quality control performed with each test run, external quality assessment through proficiency testing programs, and ongoing method validation. CLIA’s 2025 updates introduced stricter proficiency testing criteria with newly regulated analytes and enhanced requirements for quality system documentation. The updates also introduced announced inspections with up to 14 days advance notice, requiring laboratories to maintain continuous inspection readiness.
Under IVDR, Class D devices require additional scrutiny through batch testing by European Union Reference Laboratories. This requirement ensures that each production batch meets established performance standards before release to market. Manufacturers must also implement post-market surveillance systems to continuously monitor device performance and promptly address any issues that arise.
Education and training ensure competency across all personnel involved in IVD testing and result interpretation. For laboratory technicians, this includes initial qualification assessment and ongoing competency evaluation. CLIA regulations require competency assessment in six areas: direct observation of routine patient test performance, monitoring recording and reporting of test results, review of intermediate test results or worksheets, direct observation of performance of instrument maintenance, assessment of test performance through testing previously analyzed specimens, and assessment of problem-solving skills.
For physicians and other healthcare providers, education focuses on understanding test characteristics, appropriate test selection, and result interpretation. This includes awareness of test limitations, understanding when confirmatory testing is needed, and recognition of results that may require additional investigation. Manufacturers and laboratories share responsibility for providing this education, though implementation varies widely across healthcare settings.
Communication systems facilitate rapid coordination between laboratories and clinical departments when abnormal values or unexpected results occur. This includes established protocols for critical value reporting, mechanisms for clarifying ambiguous orders or results, and processes for addressing technical issues that may affect result interpretation. The 2025 CLIA updates emphasize the importance of these communication pathways, particularly as laboratories face increased complexity in testing methodologies and regulatory requirements.
Summary: Confronting Invisible Risks
IVDs do not directly contact patients, making their risks less visible and difficult to evaluate. However, the impact of incorrect test results can sometimes be more serious than that of direct-contact medical devices. The evolution of risk-based classification systems under IVDR and enhanced regulatory oversight under recent CLIA updates reflect growing recognition of these invisible but critical risks.
Responding to this specialized risk model requires not only manufacturers providing high-quality products but also laboratory technicians’ professional expertise, physicians’ understanding of testing, and patients’ appropriate awareness of testing limitations. Only when all four stakeholders understand their respective roles and work in close coordination can IVD risks be minimized and benefits maximized.
As medical care becomes increasingly sophisticated, the importance of testing in diagnosis continues to grow. The global regulatory landscape continues to evolve in response to these challenges. In Europe, the IVDR transition period extends through 2027 for Class B devices, requiring manufacturers to adapt to risk-based classification systems fundamentally different from the previous list-based approach. In the United States, the phaseout of enforcement discretion for LDTs represents a major shift in regulatory philosophy, bringing previously unregulated laboratory tests under FDA oversight while maintaining CLIA requirements for laboratory operations.
Understanding and appropriately managing the unique characteristics of IVD risk models forms an important foundation supporting the quality and safety of modern healthcare. This foundation rests on several pillars: robust regulatory frameworks that appropriately categorize risks and require corresponding controls; quality management systems that ensure consistent device performance; competent personnel at all levels of the testing process; clear communication of performance characteristics and limitations; and effective collaboration among all stakeholders.
The future of IVD regulation will likely continue evolving as technologies advance and new diagnostic modalities emerge. Artificial intelligence and machine learning are increasingly incorporated into diagnostic algorithms, presenting new challenges for regulatory frameworks designed for traditional testing methods. Point-of-care testing continues expanding, bringing sophisticated diagnostics closer to patients while raising questions about quality control and operator competency. Personalized medicine approaches demand increasingly complex tests targeting individual genetic profiles or disease characteristics.
These developments underscore the continuing importance of the fundamental principles underlying IVD risk management: recognizing that invisible risks require visible controls, understanding that indirect harms can be as serious as direct injuries, and maintaining vigilance across all stakeholders to protect patient safety. As we move forward, the regulatory frameworks governing IVDs must balance innovation with safety, accessibility with quality, and flexibility with standardization. Success in managing IVD risks ultimately depends on all stakeholders—manufacturers, laboratories, clinicians, and patients—understanding their interdependence and working collaboratively toward the shared goal of accurate, reliable diagnostic information that supports optimal patient care.
Related Articles
- Public Comments Solicitation on the Proposed Ordinance Amending the Ordinance Regarding Standards for Manufacture Control and Quality Control of Medical Devices and In Vitro Diagnostic Devices
- Understanding Risk in Pharmaceuticals and Medical Devices
- What Are the Essential Principles for Medical Devices (Japan)?
- The Latest Timeline and Implementation Status of the Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR)
- Evolution of Risk Management in Medical Devices: The Significance of ISO 14971 Revision
- Safe Design of Medical Devices: Understanding Risk Management Fundamentals
{kind=link}