

FDAは1978年以来、cGMPを品質マネジメントの国際規格であるISO-9001に整合させてきた。FDAが2006年9月に発行した「医薬品cGMPにおける品質システムからのアプローチ(Quality Systems Approach to Pharmaceutical CGMP Regulations)は、ISO-9001が標準とする品質システム(QS:品質管理システム(QMS)と同義)をもとにcGMPとの整合や、医薬品製造における品質システムの在り方を解説している。
これまで医薬品業界では、QMS(品質管理システム)を構築してこなかった。
ICH-Q10はまさしくISO-9001の概念に則って、医薬品企業全体における品質システムの構築を要求している。
FDAは1978年以来、cGMPを品質マネジメントの国際規格であるISO-9001に整合させてきた。FDAが2006年9月に発行した「医薬品cGMPにおける品質システムからのアプローチ(Quality Systems Approach to Pharmaceutical CGMP Regulatio…
一般にQMS文書体系は、以下のような構造となる。
一番上位文書は品質マニュアルである。
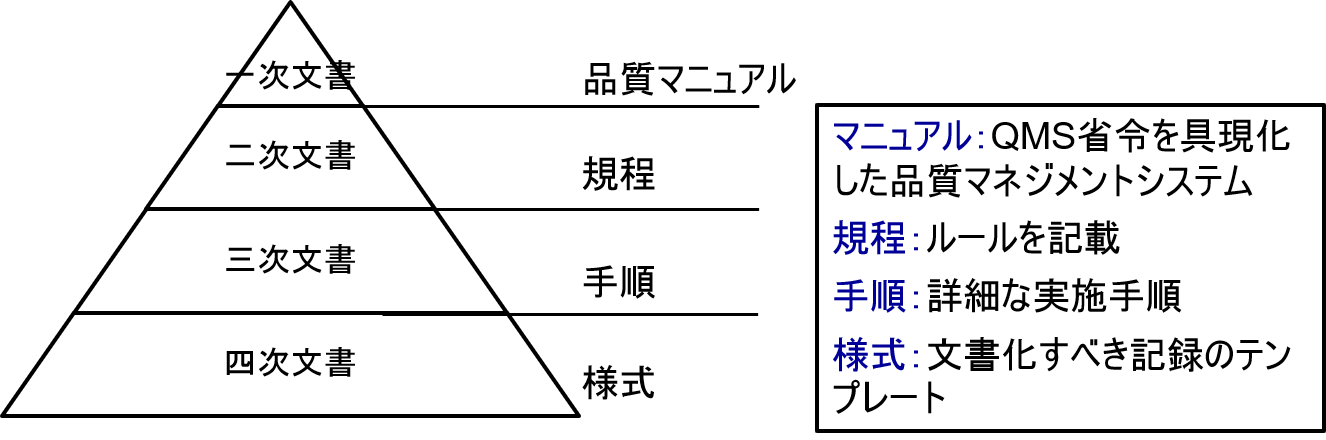
品質マニュアルでは、GMPだけではなく、探索、非臨床試験(GLP他)、臨床試験(GCP)、製造(GMP)、配送(GDP)を含めた医薬品企業全体の品質保証に関して網羅していなければならない。
通常、品質マニュアルは経営者が作成する。経営者は規制要件を遵守し、各プロセスにおける品質管理、品質保証の仕組みについて品質マニュアルにその方針を記載するのである。
なぜ同じ規制要件を参照していながら、各社によって品質マニュアルが異なるかというと、製造販売している製品が異なるためである。
製品が異なればリスクが異なる。品質マニュアルは当該医薬品のリスクに応じて適切なレベルで作成しなければならない。
いたずらに厳しいものにしてしうとコンプライアンスコストを過剰にかけてしまい、コストは薬価に乗り、結果的に患者負担となってしまうためである。
またプロセスが異なってもリスクが異なる。医薬品企業においては、製造のみを実施している場合がある(製造業)。またさらに外資系企業の場合は、二次包装のみ実施していることもある。
総合的に研究開発から製造・配送まで実施している企業も多い。
品質マニュアルはそれらプロセスを網羅し、リスクに応じた品質管理・品質保証の仕組み(品質システム)が記載されていなければならない。
品質マニュアルに従って、規程、手順書、様式等が作成される。このような文書体系のことをQMSと呼ぶ。
多くの場合、これまで医薬品業界では品質マニュアルはなく、手順書と様式のみで活動をしてきた。
これでは、適正な品質管理・品質保証が実施できないのである。
PMDAのホームページには、品質マニュアルの参考例が掲載されている。
この例では、医薬品製造(GMP)のみが対象とされており、また医薬品製造(GMP)全体を網羅していない。これでは品質システムを構築しようがない。
品質マニュアルは、品質システム構築の拠所とならなければならないのである。
{kind=link}
When someone writes an piece of writing he/she retains
the plan of a user in his/her mind that how a user can know
it. Therefore that’s why this post is perfect.
Thanks!
I needed to thank you for this excellent read!! I absolutely loved
every little bit of it. I have got you bookmarked to look at new stuff you post…