
Limitations of Sterility Testing
Birth of the Sterilization Validation Concept
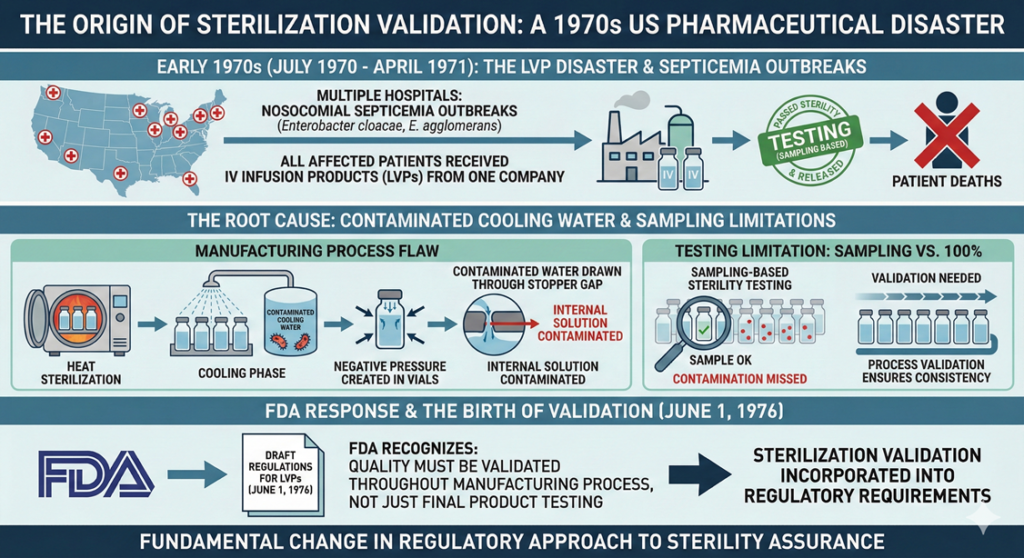
The concept of sterilization validation was born from a pharmaceutical disaster involving large volume parenterals (LVPs) that occurred in the United States in the early 1970s. This event fundamentally changed how regulatory authorities approach sterility assurance.
Between July 1970 and April 1971, multiple hospitals across the United States experienced outbreaks of nosocomial (hospital-acquired) septicemia caused by Enterobacter cloacae and Enterobacter agglomerans. All affected hospitals used intravenous infusion products manufactured by one company, and all patients who developed septicemia had received these infusion products. The investigation revealed that the products had undergone sterilization processing during manufacturing and had passed sterility testing before release.
The manufacturer had confirmed compliance with sterility testing requirements and released the products. However, patients who received these products subsequently died in a series of tragic incidents. The root cause was traced to contaminated water used as cooling water in the manufacturing process. After heat sterilization, the vials were in a reduced pressure state. This negative pressure allowed contaminated water to be drawn through the gap between the vial and rubber stopper, contaminating the internal solution.
The release testing employed was sterility testing based on sampling, not testing of every single unit. As a result, the presence of contaminated vials went undetected. This incident prompted the US Food and Drug Administration (FDA) to recognize that ensuring quality requires not only focusing on the final product but also validating that quality is maintained throughout the manufacturing process. Consequently, the FDA incorporated the concept of sterilization validation into regulatory requirements, with draft regulations for LVPs first published on June 1, 1976.
What is Sterility?
Sterilization is a process for achieving sterility—that is, a process of killing or removing all microorganisms. Sterility means the absence of all viable microorganisms.
The death characteristics of microorganisms follow an exponential function. Therefore, the presence of viable microorganisms is expressed as a probability, and even when this probability is reduced to a very low value, it never reaches zero. For this reason, sterilization operates as a probabilistic concept.
A Sterility Assurance Level (SAL) is established in advance, and sterilization conditions to achieve the SAL are calculated and implemented based on the number and types of microorganisms on the items to be sterilized (bioburden) and their D-value (Decimal Reduction Time or decimal reduction dose).
The Concept of Sterilization
Currently, a SAL of 10⁻⁶ (one in one million) is internationally adopted, and the same concept has been adopted in the Japanese Pharmacopoeia as “terminal sterilization.” This means that after sterilization, the probability of microorganisms surviving on the sterilized item is one in one million.
Understanding SAL Values
| SAL Level | Probability of Non-Sterility | Typical Application |
| 10⁻³ | 1 in 1,000 | Medical devices for intact skin contact only |
| 10⁻⁴ | 1 in 10,000 | High-level disinfection for heat-resistant medical instruments |
| 10⁻⁶ | 1 in 1,000,000 | Parenteral drugs, implantable devices, products contacting sterile tissue (Industry Standard) |
The Concept of Sterility Assurance
In general, microorganisms decrease exponentially. Probabilistically, the presence of microorganisms can never become “zero.” Therefore, a “Sterility Assurance Level (SAL 10⁻ⁿ)” was established, introducing a probabilistic concept.
As a principle, “Sterility Assurance Level (SAL) 10⁻⁶” has been defined as “sterile.” The “Sterility Assurance Level (SAL) 10⁻⁶” indicates that the probability of microorganisms surviving in a sterilized product is one in one million or less.
Sterility assurance means that the sterilization process, for the purpose of manufacturing the target product, achieves a sterility assurance level of 10⁻⁶ or better based on specific and verifiable principles.
However, sterility testing can only assure up to SAL 10⁻². To assure SAL 10⁻⁶, which is the definition of sterilization, using sterility testing would require testing one million products.
The 18th revision of the Japanese Pharmacopoeia describes sterility testing as follows:
“The sterility test is applied to active pharmaceutical ingredients or preparations that are required to be sterile. Even if results conforming to this test are obtained, this merely indicates that contaminating microorganisms were not detected in the specimens examined under the test conditions.”
In other words, it demonstrates that “passing the sterility test ≠ proof of sterility.”
FDA Warning Letter Case Study
In 2016, an FDA inspection was conducted at a Japanese ophthalmic drug manufacturer. During this inspection, deficiencies in sterilization validation and other areas were identified, and FDA Form 483 was issued.
In response to the FDA, the ophthalmic drug manufacturer stated that only lots that passed sterility testing were exported to the United States. This response ultimately became problematic, and a Warning Letter was issued.
Key Points from the Warning Letter
The FDA’s position emphasized several critical points:
- Sterilization is a Special Process: Since sterility cannot be confirmed through non-destructive testing, sterilization validation is mandatory.
- Limitations of Sampling: Sterility testing is a sampling test and cannot provide absolute assurance of sterility across all units.
- Validation Requirements: A sterility assurance level of 10⁻⁶ or better can only be demonstrated through validation of the sterilization process based on physical and microbiological methods, not through sterility testing alone.
- Testing Limitations: Sterility testing can only assure up to SAL 10⁻². It cannot detect the extremely low levels of contamination that might exist at 10⁻⁶.
- Fundamental Misconception: “Passing the sterility test ≠ proof of sterility.” This is a fundamental principle that all manufacturers must understand.
Modern Regulatory Framework
ISO Standards for Sterilization
Current sterilization practices are governed by internationally recognized standards:
- ISO 11135: Ethylene oxide sterilization validation and routine control
- ISO 11137: Radiation sterilization of healthcare products
- ISO 17665: Moist heat sterilization validation and routine control
- ISO 14937: General requirements for characterization of sterilizing agents
- ISO 11737: Determination of bioburden and selection of sterilization doses
FDA Guidance Documents
The FDA provides comprehensive guidance on sterilization:
- Submission and Review of Sterility Information in Premarket Notification (510(k)) Submissions for Devices Labeled as Sterile: Outlines requirements for demonstrating sterility assurance
- Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing — Current Good Manufacturing Practice: Addresses aseptic processing requirements
Key Regulatory Requirements
- SAL Selection: For devices labeled as sterile, FDA recommends SAL of 10⁻⁶ unless the device is intended only for intact skin contact (10⁻³ may be acceptable).
- Validation Documentation: Comprehensive validation protocols must include:
- Complete description of the sterilization process
- Validation method used (e.g., half-cycle method, overkill method)
- Bioburden determination procedures
- Installation Qualification (IQ)
- Operational Qualification (OQ)
- Performance Qualification (PQ)
- Routine Monitoring: After validation, routine monitoring must include:
- Biological indicator testing
- Physical parameter monitoring
- Periodic revalidation
- Change control procedures
Understanding the Mathematics of Sterilization
Exponential Inactivation Kinetics
Microbial inactivation follows first-order kinetics, expressed as:
N = N₀ × 10⁻ᵗ/ᴰ
Where:
- N = number of surviving microorganisms
- N₀ = initial number of microorganisms (bioburden)
- t = exposure time
- D = D-value (time or dose required for 90% reduction)
Log Reduction Requirements
To achieve SAL 10⁻⁶ from a bioburden of 10⁶ microorganisms requires a 12-log reduction:
| Stage | Population | Log Reduction |
| Initial | 1,000,000 (10⁶) | 0 |
| After 1D | 100,000 (10⁵) | 1 |
| After 2D | 10,000 (10⁴) | 2 |
| After 3D | 1,000 (10³) | 3 |
| After 6D | 1 (10⁰) | 6 |
| After 12D | 0.000001 (10⁻⁶) | 12 |
This 12-log reduction is the “overkill” approach commonly used for terminal sterilization validation.
Why Sterility Testing Alone is Insufficient
Statistical Limitations
To demonstrate SAL 10⁻⁶ through sterility testing alone would require:
- Testing approximately one million samples
- Finding zero positives
- This is neither practical nor economically feasible
In practice, sterility testing typically involves:
- Testing 20-40 units per lot (per pharmacopoeia requirements)
- This level of sampling can only provide confidence to approximately SAL 10⁻²
Practical Example
Consider a batch of 100,000 units:
- Standard sterility test: 20 units tested
- Coverage: 0.02% of the batch
- If contamination rate is 1 in 1,000 (SAL 10⁻³), probability of detection is only ~2%
- If contamination rate is 1 in 1,000,000 (SAL 10⁻⁶), probability of detection is essentially zero
Modern Approach to Sterility Assurance
Sterility assurance is achieved through a comprehensive quality system that includes:
1. Process Validation
- Establishing critical process parameters
- Demonstrating reproducibility
- Challenging the process with worst-case conditions
- Using biological indicators with known resistance
2. Environmental Control
- Classified cleanroom environments
- HEPA filtration systems
- Differential pressure control
- Environmental monitoring programs
3. Equipment Qualification
- Installation Qualification (IQ)
- Operational Qualification (OQ)
- Performance Qualification (PQ)
- Preventive maintenance programs
4. Personnel Training
- Aseptic technique training
- Gowning qualification
- Media fill simulations
- Ongoing competency assessment
5. Quality Control Testing
- Sterility testing as a release test (not for validation)
- Endotoxin testing for parenteral products
- Container closure integrity testing
- Particulate matter testing
6. Continuous Monitoring
- Routine biological indicator testing
- Physical parameter monitoring
- Trending and statistical analysis
- Change control and revalidation
Recent Regulatory Focus Areas
Contamination Outbreaks
Recent incidents highlight the critical importance of proper sterilization validation:
- 2023 Eye Drop Outbreak: Multiple brands of eye drops were recalled due to contamination with drug-resistant Pseudomonas aeruginosa, resulting in deaths, vision loss, and eye removal surgeries.
- Global Pharma Healthcare (India): FDA inspection revealed inadequate sterile manufacturing processes, lack of validated sterilization, and absence of aseptic techniques.
- 2022 Abbott Formula Contamination: Cronobacter contamination led to infant deaths and a national formula shortage, demonstrating that even established manufacturers must maintain vigilance.
FDA Enforcement Actions
The FDA has intensified enforcement related to sterility:
- Multiple Warning Letters issued to ophthalmic manufacturers for sterilization validation failures
- Import alerts preventing non-compliant products from entering the US
- Emphasis on understanding that “sterility testing passage does not equal sterility proof”
Practical Implications for Manufacturers
Common Misconceptions to Avoid
- “Our products pass sterility testing, so they are sterile”
- Reality: Sterility testing is a release test with limited sampling power
- “We only need to validate once”
- Reality: Revalidation is required after significant changes and periodically
- “Environmental monitoring is optional”
- Reality: Environmental monitoring is essential for early detection of contamination risks
- “Sterility testing can validate the sterilization process”
- Reality: Process validation requires comprehensive scientific studies, not just sterility testing
Best Practices
- Robust Validation Programs
- Use worst-case scenarios
- Include all product configurations
- Document everything thoroughly
- Involve qualified personnel
- Risk-Based Approach
- Identify critical control points
- Assess failure modes
- Implement preventive controls
- Monitor effectiveness continuously
- Quality Culture
- Train personnel comprehensively
- Encourage reporting of deviations
- Investigate all anomalies
- Implement corrective and preventive actions (CAPA)
- Regulatory Compliance
- Stay current with guidance documents
- Engage with regulatory authorities early
- Prepare for inspections proactively
- Maintain audit-ready documentation
Conclusion
The concept of sterility assurance has evolved significantly since the 1970s LVP crisis. Modern regulatory frameworks recognize that:
- True sterility is probabilistic, not absolute
- SAL 10⁻⁶ is the international standard for products labeled as sterile (with exceptions for specific applications)
- Sterility testing alone cannot validate the sterilization process or prove sterility
- Comprehensive validation combining physical, chemical, and microbiological methods is essential
- Ongoing monitoring and control ensure continued sterility assurance
Sterility assurance is maintained at a level far exceeding what sterility testing alone can guarantee. This is achieved through validated sterilization processes, environmental controls, personnel training, equipment qualification, and comprehensive quality systems. Manufacturers must understand that passing a sterility test is merely one component of a much larger framework designed to ensure patient safety.
The regulatory emphasis on process validation rather than end-product testing reflects a fundamental shift in quality philosophy: quality cannot be tested into a product—it must be built in through robust, validated processes. This principle remains as relevant today as it was when first introduced following the tragic events of the early 1970s.
Note: This document provides general guidance on sterility testing limitations and sterilization validation concepts. Manufacturers should consult current regulatory requirements, international standards (ISO 11135, 11137, 17665), and FDA guidance documents specific to their products and processes. Requirements may vary by region, product type, and intended use.
{kind=link}
Comment