EDCを利用した臨床試験における信頼性調査対応講座 (第1回)
*万が一文中に解釈の間違い等がありましても、当社では責任をとりかねます。
本文書の改訂は予告なく行われることがあります。
CSV規制要件等の改定
1. はじめに
2009年10月19日に開催された「平成21年度GCP研修会」では、演題の一つとして「電子的に収集された臨床試験データに対する信頼性調査の留意点」と題した発表が行われた。
本発表において「EDC調査チェックリスト」の案が紹介され、信頼性調査の内容と事例に基づく留意点の説明があった。
本チェックリストは、既に医薬品医療機器総合機構(以下、PMDA)のホームページで公開されている。
ER/ES指針が、平成17年に発出されてから4年半が経ったが、いよいよ本格的なER/ES指針に基づいた査察が開始されたことになる。
本シリーズでは、3回にわたって、EDCを利用した臨床試験の信頼性調査について、その内容、課題、問題点、対応方法等を考察したい。
今回は、「電子的に収集された臨床試験データに対する信頼性調査の留意点」に関する講演内容の要約を行った。
2.平成21年度GCP研修会
平成21年度GCP研修会は、10月19日に東京、10月23日に大阪で開催された。
そのアジェンダは、図1に示すとおりである。
| 時 間 | 内 容 |
| 13:00~13:10 | 挨拶 (東京)独立行政法人 医薬品医療機器総合機構 理事(技監) 川原 章 (大阪)独立行政法人 医薬品医療機器総合機構 理事長 近藤 達也 |
| 13:10~13:40 | 治験の計画等の届出、治験中の副作用・不具合報告及びIRB登録制度について (東京)PMDA審査マネジメント部 星 順子 (大阪)PMDA審査マネジメント部 北原 淳 |
| 13:40~14:00 | 医療機器GCPの改正及び医療機器の基準適合性調査について (東京)信頼性保証部主任専門員 疋田 理津子 (大阪)信頼性保証部調査専門員 福岡 由紀 |
| 14:00~14:20 | 適合性書面調査の現状と留意点 (東京)信頼性保証部調査専門員 魚津 公一郎 (大阪)信頼性保証部調査専門員 南谷 浄 |
| 14:20~14:40 | GCP実地調査の現状と留意点 (東京)信頼性保証部調査専門員 田中 紀子 (大阪)信頼性保証部調査専門員 田尻 興保 |
| 14:40~15:10 | 治験を行う実施医療機関における留意点 (東京)信頼性保証部調査専門員 城谷 真理 (大阪)信頼性保証部調査専門員 山本 健児 |
| 15:30~16:00 | 電子的に収集された臨床試験データに対する信頼性調査の留意点 (東京)信頼性保証部調査専門員 宇井 英明 (大阪)信頼性保証部調査専門員 福島 弘子 |
| 16:00~16:20 | 医療用後発医薬品に係るGCP実地調査の留意点 (東京)信頼性保証部調査専門員 今泉 克明 (大阪)信頼性保証部調査専門員 川名 純一 |
| 16:20~16:50 | GPMSP/GPSP調査の現状と留意点 (東京)信頼性保証部主任専門員 森山 祐輔 (大阪)信頼性保証部調査専門員 松原 芳幸 |
| 16:50~17:00 | 挨拶 財団法人 日本薬剤師研修センター 専務理事 平山 一男 |
図1 平成21年度GCP研修会アジェンダ
研修会の中で、「電子的に収集された臨床試験データに対する信頼性調査の留意点」という演題があり、EDCを利用した臨床試験における信頼性調査の内容と事例に基づく留意点の説明があった。
本講演の冒頭で「EDC調査に関する検討の位置づけ」として、PMDAの平成21年度計画の紹介があった。(図2)
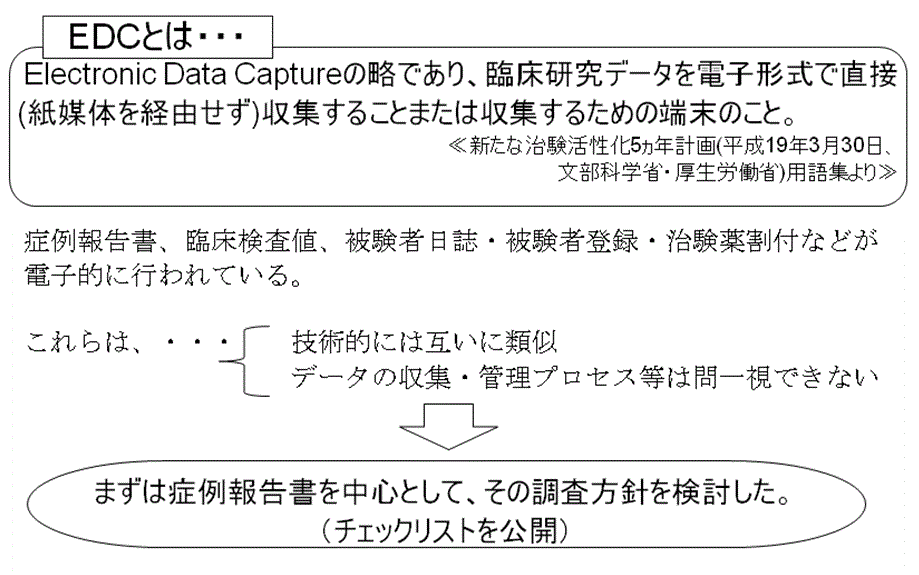 図2 EDC調査に関する検討の位置づけ
図2 EDC調査に関する検討の位置づけ
PMDAの中期計画及び21年度計画において、「急速に進んでいる治験の電子化に対応するため、EDCシステムを中心にシステム調査の検討を進める」とある。
EDCに関する調査において、まず確認すべき事項を定義し、次にシステム調査で確認が可能かどうか、困難をともなうかどうかを検討したとのことであった。
続いて、本講演では、以下の3項目について説明があった。
1) 医療機関で収集される臨床試験データについて
2) 臨床試験データを電子的に取り扱うための関連法令等
3) 電子的に収集されたデータの基準適合性調査と留意点
3. 医療機関で収集される臨床試験データについて
本講演の一番目の内容は、「医療機関で収集される臨床試験データについて」であった。
新たな治験活性化5ヵ年計画の用語集には、EDCとは「Electronic Data Captureの略であり、臨床研究データを電子形式で直接(紙媒体を経由せず)収集することまたは収集するための端末のこと。」とある。(図2 参照)
本発表によると、この用語の定義が意味するところは、かなり広範囲であるという。
例えば、治験依頼者が医療機関から臨床試験データを取得する場合を考えても、症例報告書、中央検査機関による臨床検査値、被験者日誌・被験者登録・治験薬割付など、多くの電子化が行われている。
しかしながら、これらは技術的には互いに類似してはいるものの、データの収集・管理プロセス等は必ずしも同一のものではない。
まずは症例報告書を中心として、その調査方針を検討し、チェックリストを公開したとのことであった。
4. 臨床試験データを電子的に取り扱うための関連法令等
本講演の二番目の内容は、「臨床試験データを電子的に取り扱うための関連法令等」であった。
EDCを利用したとしても、症例報告書の作成については、薬事法施行規則第43条、GCP省令(第47条等)を遵守する必要がある。このことは症例報告書以外のデータでも同様である。
すなわち、基準適合性調査を実施する上での基本的な考え方は変わるものではない。
それらに加えて、電子記録を利用するのであれば、GCP第26条、ER/ES指針およびその上位法令にも留意する必要がある。
4.1 GCP省令第47条 症例報告書等
GCP省令第47条には、症例報告書の作成、署名、修正に関しての規定が記載されている。(図3 参照)
| (症例報告書等) 第47条 治験責任医師等は、治験実施計画書に従って正確に症例報告書を作成し、これに記名なつ印し、又は署名しなければならない。 2 治験責任医師等は、症例報告書の記載を変更し、又は修正するときは、その日付を記載して、これになつ印し、又は署名しなければならない。 3 治験責任医師は、治験分担医師が作成した症例報告書を点検し、内容を確認した上で、これに記名なつ印し、又は署名しなければならない。 |
図3 GCP省令 第47条
治験責任医師等は、症例報告書を作成した際には、記名・捺印または署名を行わなければならない。
また症例報告書の記載を変更したり、修正する際には、その日付を記載したうえで、捺印または署名を行わなければならない。
さらに、治験分担医師が作成した症例報告書であった場合、最終的に治験責任医師がその内容(たとえば、記載事項や修正が適切か等)を確認したうえで記名・捺印または署名を行わなければならない。
また運用通知では、治験責任医師は症例報告書の写しを保存しなければならないとされている。(図4 参照)
| 治験責任医師又は治験分担医師は、症例報告書を治験実施計画書の規定に従って作成し、記名捺印又は署名の上、治験依頼者による治験においては治験依頼者に提出し、自ら治験を実施する者による治験においては自ら治験を実施する者が保存しなければならない。また、治験依頼者に提出した症例報告書の写しを保存するものとする。 |
図4 GCP運用通知 第47条 第1項
4.2 GCP省令第26条 記録の保存等
GCP省令第26条には、記録の保存等に関する規定が記載されている。(図5 参照)
| (記録の保存等) 第26条 治験依頼者は、次に掲げる治験に関する記録(文書及びデータを含む。)を被験薬に係る医薬品についての製造販売の承認を受ける日(第24条第3項の規定により通知したときは、通知した日後3年を経過した日)又は治験の中止若しくは終了の後3年を経過した日のうちいずれか遅い日までの期間適切に保存しなければならない。 1) 治験実施計画書、契約書、総括報告書その他この省令の規定により治験依頼者が作成した文書又はその写し 2) 症例報告書、第32条第6項の規定により通知された文書その他この省令の規定により実施医療機関の長又は治験責任医師等から入手した記録 3) モニタリング、監査その他の治験の依頼及び管理に係る業務の記録(前2号及び第5号に掲げるものを除く。) 4) 治験を行うことにより得られたデータ 5) 第16条第5項に規定する記録 |
図5 GCP省令 第26条
このなかで、治験依頼者は症例報告書を保存しなければならないことが明記されている。
また運用通知において、「記録の保存等 第26条第1項」にかかわる事項として、電子データ処理システムに関する要件があげられている。(図6 参照)
| 3 治験依頼者は、データの処理に当たって、電子データ処理システム(遠隔操作電子データシステムを含む。)を用いる場合には、次の事項を実施しなければならない。電子データ処理システムが、完全性、正確性、信頼性及び意図された性能についての治験依頼者の要件を満たしていることを保証し、文書化すること(すなわちバリデーションされること。)。当該システムを使用するための手順書を整備すること。当該システムが、入力済みのデータを消去することなしに修正が可能で、データ修正の記録をデータ入力者及び修正者が識別されるログとして残せる(すなわち監査証跡、データ入力証跡、修正証跡が残る)ようにデザインされていることを保証すること。データのセキュリティ・システムを保持すること。データのバックアップを適切に行うこと。データの修正を行う権限を与えられた者の名簿を作成し、管理すること。盲検化が行われている場合には、盲検性が保持されるようにすること。 |
図6 GCP運用通知 記録の保存等 第26条第1項
これらは、ICH-GCPの邦訳であり、平成9年3月27日に中薬審答申第40号として発表されたものと同一である。
このなかで「遠隔操作電子データシステム」とは、EDCシステムを指すと考えられる。
1) バリデーション
バリデーションは、コンピュータシステムを使用する際の必須要件である。
2) 手順書
当該システムを使用するための手順書を整備しなければならない。手順書は、システムの操作手順書のみではなく、セキュリティ管理に関する手順書、監査証跡確認手順書、電磁的記録媒体の保存性に関する手順書、バックアップ/リカバリ手順書等の、電子記録・電子署名を使用する上で必要となる手順書を含む。
3) 監査証跡
当該システムは、監査証跡が自動的に記録されるように設計されていなければならない。
監査証跡は、改ざんを発見するために必須の機能である。
4) セキュリティ
データのセキュリティ・システムを保持しなければならない。
セキュリティは、なりすましや改ざんを防止するために必要である。
セキュリティには、物理的セキュリティ、論理的セキュリティ、ネットワークセキュリティ、人的セキュリティなどが存在する。
物理的セキュリティは、サーバルームには施錠を行い、無用な者を入室させないというものがあげられる。またEDCシステムの端末を、往来の多い場所を避け、研究室などの施錠できる場所に設置するなども考えられる。
論理的セキュリティとは、パスワードを利用し、アクセス制限や権限設定を行うことを指す。また一定時間入力がなかった場合に、自動的にスクリーンセーバーなどを起動させることも有効である。
ネットワークセキュリティは、ファイヤーウォールやウィルス駆除ソフトウェアなどの導入を指す。またウィニー(Winny)などの不適切なソフトウェアがインストールされている場合、情報漏えいにつながる危険性がある。
人的セキュリティは、パスワードを他人に教えない、紙などに記載しておかないなどである。これらを周知徹底するために、ユーザ教育は重要である。
5) バックアップ
既に解説した通り、バックアップは規制要件であり、真正性の要件である。
データのバックアップを適切に行うことも必要であるが、災害時等に適切に回復(リカバリ)できることも重要である。
6) データの修正を行う権限を与えられた者の名簿
データの修正を行う権限を与えられた者は、電子記録の改ざんが可能になる。したがって、適宜、誰が修正権限を持っているのかを適切に管理しなければならない。
年度初めなどには、治験責任医師等の異動が多くなる。当該治験責任医師等が、当該治験を外れた場合、ユーザ権限をすみやかに無効化しなければならない。
また最終の被験者が治験を終えた際(LPO:Last Patient Out)の際などにも、必要のなくなった権限は無効化しておかなければならない。
これらアクセス権限等を、アカウント管理表等を作成し、常時管理しなければならない。
7) 盲検性の保持
EDCシステムを利用すると、症例データの集積にともない、リアルタイムに治験中のデータの抽出や中間解析が可能となる。
盲検化された試験の場合、中間解析等を行うことは、盲検性の保持の面から極めて不適切である。
5. 電子的に収集されたデータの基準適合性調査と留意点
本講演の三番目の内容は、「電子的に収集されたデータの基準適合性調査と留意点」であり、多くの聴衆が関心を持つ内容である。
講演内容によると、平成20年度以降、10社以上(20申請品目以上)でEDCを利用した試験を含む申請品目の適合性調査がPMDAによって実施されてきたという。ちなみに平成19年度までは2申請品目だったとのことであった。
PMDAでは、平成 21年3月に、EDCデータに対する調査すべき事項を「EDC調査チェックリスト(案)」としてまとめた。
その後、平成21年5月から8月にかけて、「EDC調査チェックリスト(案)」を用いてパイロット調査を行ってきたという。
このチェックリストは、既にPMDAのHPで公開されている。
http://www.pmda.go.jp/operations/shonin/outline/shinrai/checklist.html
本チェックリストは、Wordが治験依頼者用、pdfが医療機関用となっている。
ちなみに、このようなチェックリストを課長通知等ではなく、PMDAのホームページで公開するという意義は、常に見直しと改定が行われることを意味すると筆者は理解している。
本チェックリストの内容は、図7に示す通りとなっている。
| (平成21年10月現在)EDC調査チェックリスト (治験依頼者用)1. システムの概要について 2. EDCシステム運用に関する治験依頼者の組織・体制・委託状況等 3. バリデーションについて 4. ユーザー管理について 5. データの保存 6. 電子症例報告書の作成、修正及び署名についてEDC調査チェックリスト(医療機関用)1. ユーザー管理について 2. データの保存 |
図7 EDC調査チェックリストの構成
本演題では、チェックリストの内容の紹介とともに、調査の内容と事例に基づく留意点の説明があった。
チェックリストは、本来の調査項目に加えて、EDCを使用した場合に追加となる差分の項目のみが記載されていることの説明があった。
監査証跡のバリデーションに関しての調査項目が存在することが特徴的である。
また治験依頼者の要件を満たしていることを保証する文書の調査も加わった。
さらに各種手順書に関して、当該治験に該当するものの作成日を調査することになっている。
バリデーションに関しては、ASPを使用するケースを例とし、開発者、ASP、治験依頼者の関係を図で示し、このパターンが最も複雑になること、しかしながら最終責任は治験依頼者にあることの説明があった。
バリデーションについての深い調査は行っていないが、要検討項目であることが付け加えられた。
5.1 治験依頼者に関する留意点
治験依頼者に関する留意点として、以下のような事例等が報告された。
1) ユーザ管理において、システムが万全であっても、使う側が適切に運用しなければならないこと。
2) 教育訓練を行っていても、ID、Passwordの発行時に、受け手(医療機関)が理解できていない事例が発生している。
3) データの保存に関する留意事項として、多くはpdfで作成されているが、治験責任医師の署名後に修正が発生した場合、最終版が保存されていない事例があった。
これは第26条の適切な保存の要件に抵触するものである。
4) 電子症例報告書の作成、修正および署名について、本来(紙ベースのCRFの場合)は医療機関が責任を持つべきであるが、EDCを利用する場合は、治験依頼者がそれらの環境を準備するため、治験依頼者側での調査を行っている。
医療機関においては、紙ベースの調査と変わらないため、本項目の医療機関用チェックリストは作成していない。
5) 電子症例報告書は、監査証跡を含めて完全といえる。
6) pdf化する際に特殊文字などによる文字化け、表示のズレ等が発生している事例がある。
7) EDCでは、権限設定により、エラーを防ぐことができることがある。
例えば、最終確認署名は治験責任医師のみが可能となるようにし、治験分担医師はダメとするなど。
あくまでも責任は医療機関側にあるが、色んなシステムがあり、覚えきれないので、(機能を工夫するなど)できることをやってもらいたい。
5.2 医療機関に関する留意点
医療機関に関する留意点として、以下のような事例等が報告された。
1) ID・パスワードが、医療機関毎にひとつ(共有)と勘違いしたケースがある。
2) 資格のない者に入力を代行させている事例がある。これはEDCを利用しているかどうか以前の問題である。
3) 利用権限が同等以上であるユーザであれば、代わりに入力してもかまわないといった勘違い事例があった。
4) 医療機関が電子症例報告書(写)を受け取る際、治験依頼者が作成したものであるから大丈夫であろうとする考え方は良くない。
悪意はなくとも人為的ミスはあり得る。電子症例報告書(写)を受領する際に、内容を確認すること。
記録の保存責任はあくまでも医療機関にある。受領書にその旨記載されていると思う。受領書にサインしたことによって内容に責任を持つことになる。
6. おわりに
今回は、平成21年度 GCP研修会における、「電子的に収集された臨床試験データに対する信頼性調査の留意点」に関する講演内容の要約を試みた。
本講演では、事例に基づいた留意点が紹介されたが、これはあくまでもPMDAの査察官が見付け出せた内容に基づくものである。
今後も調査が継続され、本チェックリストは事例に基づき改定されるものであろうと推察する。
しかしである。本来、規制当局は警察官のような立場であっては困る。
たまたま調査で見付け出すことができた事例に関しては、指摘を行い改善が図られるが、そうでない事項においては瑕疵が残存してしまうからである。
例えばスピード違反の場合、警察官が目撃している場合には取り締まられるが、そうでない場合には違反が見逃されることになる。指摘した場合のみ是正が行われるといった方法では、根本的な品質保証は図れない。
かつてのFDAもそうであった。1997年に成立した、FDA近代化法以降、FDAは査察方針について見直しと改善を行った。
FDAが発行する最近のワーニングレターには、次のような一節が加えられることが多い。
「このワーニングレターは、貴社におけるすべての違反を列記したものではないことを、承知して頂きたい。該当する法律やFDAの規制要件を遵守することの保証に関する責任は貴社にある。」
問題は、日本では当局がEDCに関する規制要件を発表していないことである。規制要件がなければ、遵守すべき事項が不明確である。したがって、適合性を持ったSOPの作成もできないことになる。
本来チェックリストは、拠り所とする規制要件に基づいて作成されるべきである。
GCP省令26号や、ER/ES指針では不十分である。
またCSVに関する規制要件も発表されていない中で、バリデーションを実施しているかといった調査も無理があるように思える。
このような方法では、調査がうわべだけのものになってしまう。
FDAが、2007年5月に発行した「Guidance for Industry Computerized Systems Used in Clinical Investigations」では、背景として次のように述べている。
「FDAが臨床試験データを受領するかどうかは、オンサイトの査察や監査中に、データの品質や完全性を確認できるかどうかというFDAの能力にかかっている。」
奇しくも、平成19年12月21日に日本製薬工業協会 医薬品評価委員会が開催した「臨床試験データの電子的取得に関するガイダンス」説明会において、PMDA 新薬審査第2部(当時)の井本昌克氏が講演し、「EDCを利用した試験成績が受け入れ可能か不明」と述べている。
次回は、「EDC調査チェックリスト(案)」の内容を筆者なりに解釈し、製薬企業における対応の方法等について考察を行ってみたい。
参考
1) 「平成21年度 GCP研修会 講演要旨集」 平成21年10月19日 財団法人 日本薬剤師研修センター
2) 「医薬品等の承認又は許可等に係る申請等における電磁的記録及び電子署名の利用について」平成17年4月1日 薬食発第0401022号
3) 「臨床試験データの電子的取得に関するガイダンス」平成19年11月1日 日本製薬工業協会 医薬品評価委員会

EDCを利用した治験講座 その1
EDCを利用した治験講座 その2
EDCを利用した治験講座 その3
{kind=link}
Comment