
内容を読んでいると現状ではどの企業でも遵守できていないような内容や遵守困難な内容が含まれていることがある。
これは、欧米の規制要件が期待を述べているためである。
規制要件のあるべき姿は、業界を指導し理想的な状態に引っ張っていくことである。
したがって、現状では遵守できていなくとも、2~3年後にはそうなっていて欲しいという期待値が記載されているのである。
つまり規制要件が改定されたからといって、即座に不遵守が指摘され行政処分を受けることはまずないのである。
ただし、何年も旧態依然とした体制やQMSの状態で放置していればいずれ指摘を受けることになるだろう。
PIC/S GMPをはじめ、FDAやMHRAなどは先進的な規制要件を発出している。内容を読んでいると現状ではどの企業でも遵守できていないような内容や遵守困難な内容が含まれていることがある。これは、欧米の規制要件が期待を述べているためである。規制要件のあるべき姿は、業界を指導し理想的な状態に引っ張って…
一方で、日本の規制要件は多くの場合、後追いである。
現状で多くの企業が遵守できていそうな内容や遵守するために困難ではない内容を記載しているように見受けられる。
したがって、欧米の規制要件に比べて数年遅れた内容になっていることが多い。
しかも、日本の規制要件の構成は実に複雑である、GMP省令、施行通知、課長通知、Q&A、事例集など多岐にわたる。
いったいどれが規制要件としての必須事項であろうか。
ともするとQ&Aに大事なことが記載されている場合がある。
また、本邦においてはPIC/S GMPは課長通知の扱いであり、省令ではない。
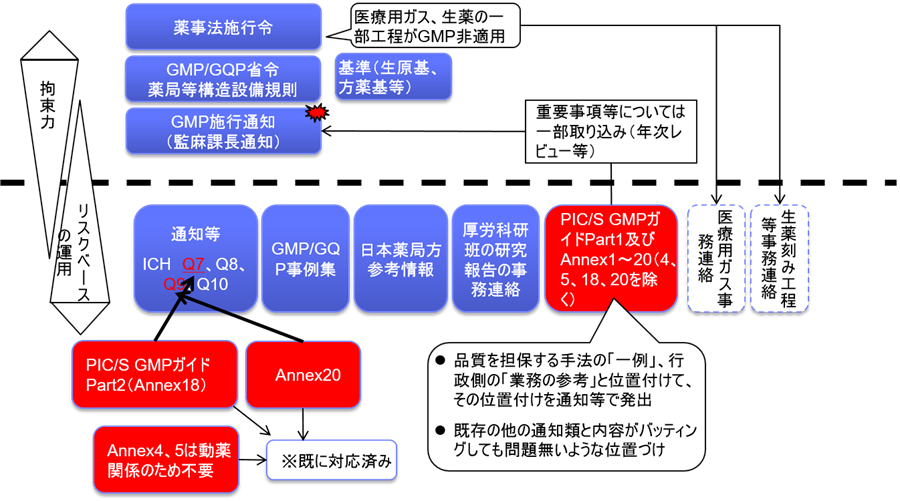
話は変わるが、PIC/S は規制当局の集まりである、したがって、PIC/S GMPは規制当局の査察官に対して発出されている。
つまり、査察官が査察時にチェックしなければならない事項が記載されているのである。
製薬企業が手順書に落とす場合はその点を理解(査察官向けであるということ)しながら作業しなければならない。
{kind=link}
Comment