

*万が一文中に解釈の間違い等がありましても、当社では責任をとりかねます。
本文書の改訂は予告なく行われることがあります。
*万が一文中に解釈の間違い等がありましても、当社では責任をとりかねます。 本文書の改訂は予告なく行われることがあります。…
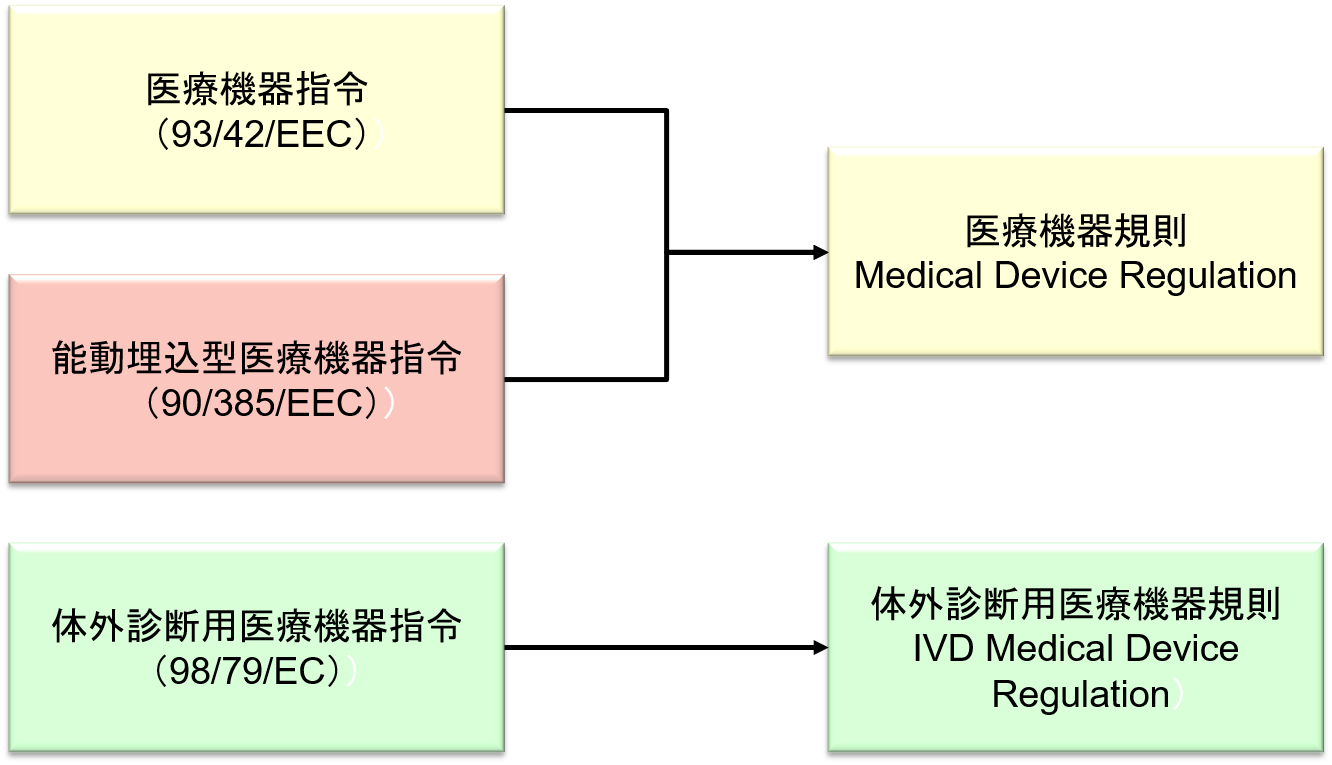
2017年5月5日に、医療機器指令 Medical Device Directive (93/42/EEC)が改正され 「欧州医療機器規則 (MDR:Medical Device Regulation)」 が公示された。
20日後の2017年5月25日より施行され、2020年5月25日までの3年間が移行措置期間とされている。Manufacturer は、この移行期間中に技術文書を改訂し、新しい要求事項に対応しなければならない。
欧州では約20年ぶりの大改編である。
この改正内容は多岐に渡り、また上市済の製品の対策も必要となるため、強制化に間に合うように計画的に対策を進める必要がある。
MDRは、従来のMDDと能動埋め込み型医療機器指令 Active Implantable Medical Device Directive (90/385/EEC)を統合し置き換えるものである。
MDRは条文が難解で非常にわかりにくいのが難点である。
MDRは、日本や米国の法規制を参考にして作成され、世界で最も厳しい規制となった。
MDRは、技術文書について、従来よりも踏み込んだ精査を求めている。
臨床評価と市販後臨床フォローアップについてより厳しい要求事項となった。
サプライチェーンを通じた機器のより良いトレーサビリティを求めることで、品質の安全性と性能に関する評価に対する懸念に対処している。
指令(Directive)から規則(Regulation)へ:背景
2012年、フランスのPIP(ポリ・アンプラン・プロテーズ)社が工業用グレードのシリコンを医療用シリコンと偽り販売していたことが発覚した。
PIP製の豊胸バッグが最初に問題になったのは2010年のことである。埋め込み手術を行う医師らが、同社豊胸バッグの破裂率が異常に高いことを指摘していた。
同社のシリコンは65か国で約30万人の女性に埋め込まれたとみられ、翌2011年には世界各地で問題視された。
フランス裁判所は当該製品を認証したNBが検査義務と注意義務を怠ったと判断し、当該NBへ賠償命令を出した。
このようなインシデントを予防するために指令の変更を行う必要性が話し合われた。
その結果、システムに対する早急な変更が提案された。
- 市販後監視の強化
- 通常監査に追加でunannounced visitsの実施
- トレーサビリティの確保
指令(Directive)と規則(Regulation)の違い
規則(Regulation)
- 加盟国に対し、国内法への適用を待たずに直接拘束力を有する。
指令(Directive)
- 加盟国を拘束するが、その具体的な形式及び手法は加盟国に委ねられる。
- 適用にあたっては、加盟国内での実施手続(担保法の制定等)が必要。
MDRの目的
- 医療機器および体外診断薬の安全性の確保
- 革新的なヘルスケアへの患者のタイムリーなアクセス
MDR/IVDRの経過
- 2012年MDR/IVDRのProposal発行
- 2013年、MDR/IVDRのドラフト発行
- 2016年6月、EU CouncilおよびEU議会が新規制に合意
- 2016年9月、MDR/IVDRのドラフトを全加盟国の公用語に翻訳
- 各言語でのMDR/IVDRのドラフトの改訂後、EU議会による新規則の承認
- 3年の移行期間を経て、MDRの発効(2020年5月)
- 5年の移行期間を経て、IVDRの発効(2022年5月)
MDRにおけるMDDからの変更点
製品の市販までのルールの強化
- 各国当局によるNBに対する監視の強化
- NBによる製造業者への非通知監査の実施
- ハイリスク製品はSpecial Notified Bodyのみが認証可能
臨床試験への要求強化
- 市販後監視の強化
- 製造業者による製品の品質、性能、安全性に関するフォローアップ責任の明確化
- 製造業者は重大な問題について、再発防止策をEUポータルを通じて報告
トレーサビリティの強化
- UDIの要求
- EUの共通データベースへの製造業者および製品情報の登録
クラス分類

{kind=link}
Comment