
どのベンダーも医療機器業界とFDA対応をうたっているようだ。
しかしながら、実際にデモをみたり、プレゼンテーションを受けてみても、本当にFDA対応ができているシステムというのは、皆無のようである。
またベンダーには、FDA対応のためのスキルを持った要員がいないことがほとんどで、逆に医療機器メーカからノウハウの提供を受けたいと願っているところばかりである。
多くのベンダーから、PLMシステムが販売されている。どのベンダーも医療機器業界とFDA対応をうたっているようだ。しかしながら、実際にデモをみたり、プレゼンテーションを受けてみても、本当にFDA対応ができているシステムというのは、皆無のようである。またベンダーには、FDA対応のためのスキルを持った要員…
ではいったいFDA対応のPLMシステムとはどういうものであろうか。
グランドデザインの重要性

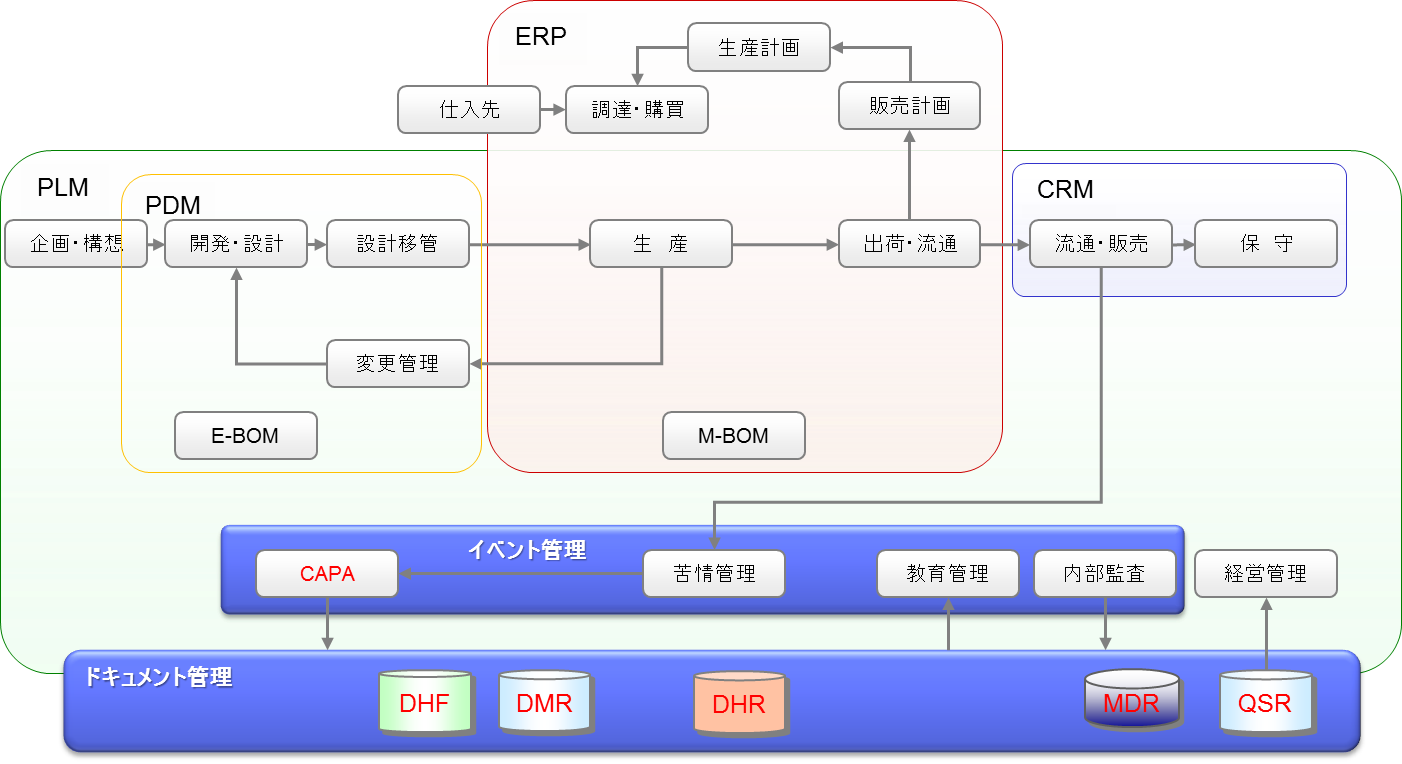
FDA規制対応が必要な医療機器業界では、一般製造業と違って、CAPA、DHF、DMR、DHR、MDR、QSRの構築が必須である。
“15分ルール”
「査察官の要求に対し、15分以内に適確な資料等を提示しなければならない。」という暗黙のルールのこと。(実際に明文化されているわけではない。)
もし資料の提示に15分以上かかる場合は、“査察コーディネータ”は、いつまでに(例:本日の午後、明日の朝など)資料を提示できるかを説明し、次の質問に移ってもらうように要請しなければならない。
15分以上、査察官を待たせるということは、「査察妨害」ととらえられる可能性がある。
待たされた査察官は、本来指摘に及ばない事項に関しても、言及することになりかねない。
紙ベースで査察に対応する場合、15 分以内に適切な記録を探し出すためには、すべての文書・記録を査察が行われている部屋の近くに配置しておかなければならない。その上で、各部署の担当者が即座に適切な文書・記録を探し出し、届けられなければならない。
文書・記録は、出来る限り電子化されていることが望ましいが、その場合は電子記録の信頼性も問われることになる。つまりPart11 対応が重要となるのである。

CAPA
非常に高い確率で調査されるのは、CAPA である。日本の企業では、CAPA が紙ベースやExcel などで管理されていることが多い。
また苦情管理のみ対象としている企業も多い。近年は、CAPA システムの構築がほぼ当たり前となりつつあり、FDAの指摘(483 フォーム)が最も出されているところである。
輸出品目とは限らない
多くの企業は、当該米国輸出品目に関する文書と記録を調査されるものと思っているかも知れない。FDA 査察では、輸出品目以外も調査されることがある。
なぜならば、FDA の査察官は、当該企業の「姿勢」に関心を持っているからである。
やってはならないことは、ダブルスタンダードである。つまり、企業内に例えば輸出用と国内用といったように、SOPを2系統作ってしまうということである。
FDAの査察の傾向
FDAの査察は、ある事象に対して“狭く深く”質問がなされる。(Mock Inspectionは、“広く浅く”質問する。)
例)ある顧客クレームに関して、
- いつ、クレームを受け取ったか
- 根本的な原因は何であったか
- 手順書(またはその他関連資料)の何を変更したか
- 変更後の手順書に関して、教育訓練をいつ、誰に実施したか

*1 PLM(Product Lifecycle Management)は、10年以上前に提唱され、企業における製品の設計・開発・保守・廃棄・リサイクルなど、製品のライフサイクル全体を通して製品関連情報をITで一元管理し、収益を最大化していくための方策のことである。
【ここがポイント】
★ 各ベンダーから発売されているPLMシステムで、FDAの医療機器規制に適合したものは皆無である。
★ 既にPDM、ERPを導入している医療機器企業が、FDA の医療機器規制に適合するための方策とは。
★ 一般製造業では、医療機器部門のためにだけPLMシステムをカスタマイズするわけにはいかない。
★ FDA規制対応のために足りない機能は、イベント管理機能、ドキュメント管理機能である。

{kind=link}
Comment