
…
From QSR to QMSR

On February 23, 2022, FDA will adopt the current QSR (Quality System Regulations: 21CFR Part 820 Quality System Regulations) to align with ISO 13485:2016proposed amendments.
The regulation to align QSRs with ISO 13485:2016 is “Quality Management System Regulations / QMSR“.
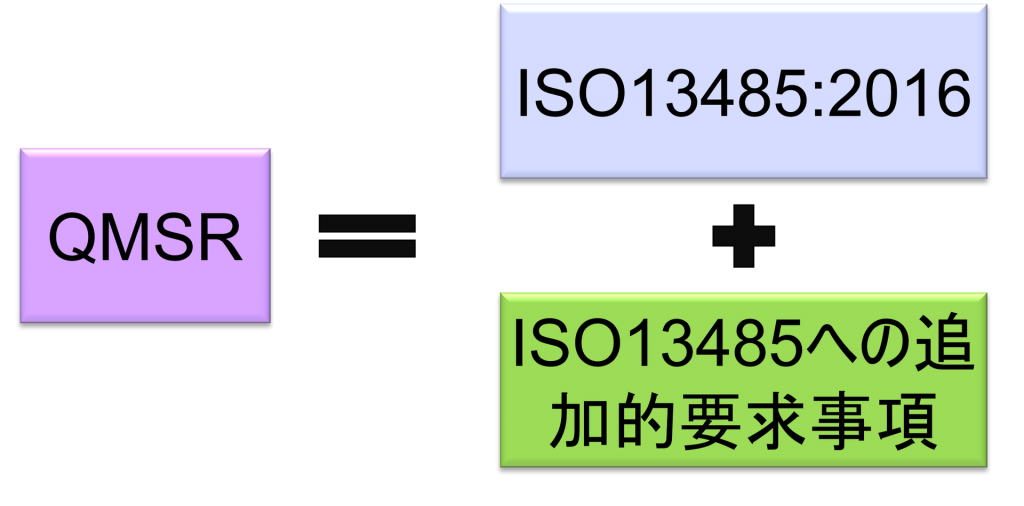
The QMSR will align many of the requirements with ISO 13485:2016 by incorporating ISO 13485:2016 requirements.
On the other hand, it adds additional requirements to ISO 13485:2016 regarding the retention of complaint files and other records.
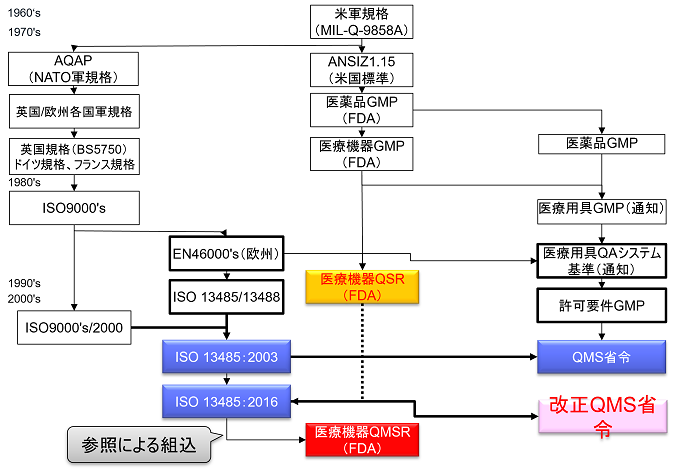
The QSR has served as the medical device quality system regulation in the United States for more than 20 years since the current rule went into effect on October 7, 1996. Throughout this time, the QSR has never been revised, although there have been minor amendments. For this QSR to QMSR revision, FDA intends to consolidate the requirements in the US with the quality management system requirements used by other regulatory authorities.
Key Points of Revision
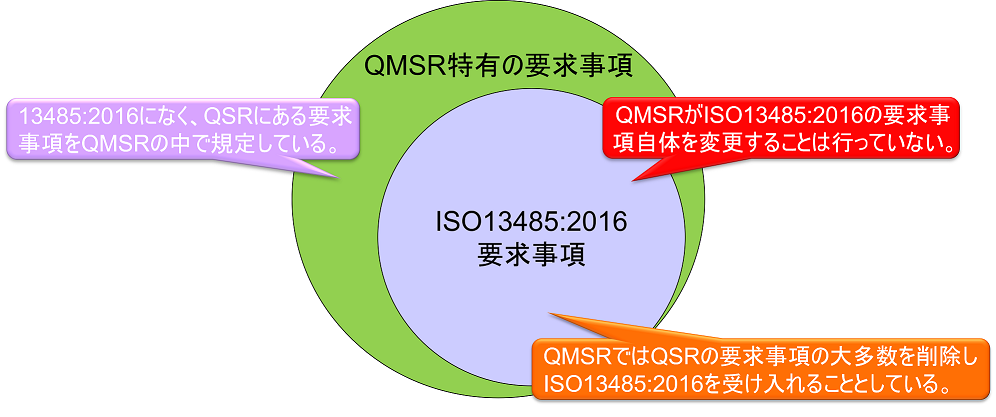
In Part 820, FDA seeks to align with QMS requirements used by other regulatory agencies by incorporating ISO 13485:2016 by reference. This amendment changes the title of the Part 820 regulations from Quality System Requirements (QSRs) to Quality Management System Requirements (QMSRs). In principle, the QMSRs delete the current Part 820 QSR requirements and incorporate the requirements of ISO 13485:2016. The QMSR did not make any changes to the requirements of ISO13485:2016 itself. While Japan incorporates the requirements of ISO 13485:2016 into the main body of the QMS regulation (Chapter 2) and specifies additional Japanese requirements in Chapter 3, the QMSR does not incorporate ISO 13485:2016 but uses it as a reference. The QMSR does not incorporate ISO13485:2016 but takes the form of a reference.
However, some definitions in the current QSRs will be retained or retained with changes. In other words, the requirements in the QSR, but not in ISO 13485:2016, are specified in the QMSR. (The requirements in ISO 13485:2016 are accepted as is, with FDA’s own requirements added.) FDA-specific requirements and provisions are also added to clarify certain concepts used in ISO 13485:2016.
QMSR requirements are virtually identical to QSR.
The requirements of the current Part 820 (QSR) are substantially the same as those of ISO 13485:2016 as a whole. Thus, the requirements of the QMSR are virtually identical to those of the QSR, as the requirements specific to the QSR are retained in the QMSR.
Key Point 1: The requirements of the QMSR are virtually identical to those of the QSR.
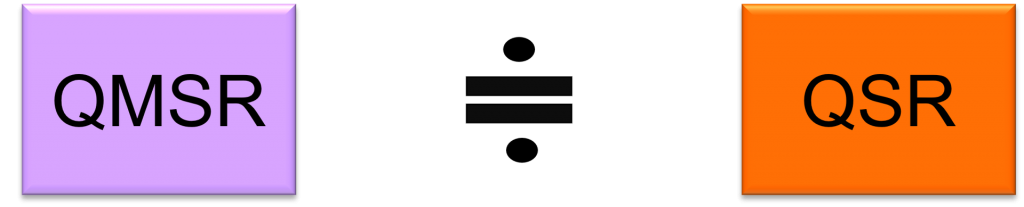
Key point 2: ISO 13485:2016 taken verbatim, with additions requirements
Key Point 3. designed to lead to compliance with QMSR = compliance with ISO 13485:2016.
Additional Requirements to ISO 13485.
In the diagram above, the QMSR-specific requirements are as follows
recordkeeping requirements (§820.35)
1. recordkeeping requirements (§820.35)
- Signature of the person approving the record and the date of approval
- Records of complaints
- Records related to ancillary services
- Records related to UDI
- Handling of Confidential Records
2. labeling and packaging requirements (§820.45)
3. UDI (see §820.10 to Part 830)
4. Traceability (see §820.10 to Part 821)
5. Adverse event reporting (see §820.10 to Part 803)
6. recall (see §820.10 to Part 806)
The following record components are not specified in the QMSR as they can be accomplished by following the provisions of ISO 13485
- Design History File (DHF) (currently 820.30(j))
- Device Master Registry (DMR) (now 820.181)
- Equipment History Register (DHR) (now 820.184)
- Quality System Records (QSR) (currently 820.186), etc.
Relation between ISO 13485 certification and FDA inspections after QMSR adoption
After the QMSR is finalized, the Quality System Inspection Technique (QSIT) will be reviewed. An FDA inspection will not result in the issuance of an ISO 13485 certificate. Having an ISO 13485:2016 certificate of conformity does not exempt you from an FDA inspection. The FDA will not develop an ISO 13485 certification program. Although the QMSR will incorporate ISO 13485, the FDA will not grant any burden reduction if a company has ISO 13485 certification.
関連商品
[blogcard url=”https://ecompliance.co.jp/SHOP/O094.html” title=”【VOD】QMSRセミナー (Quality Management System Regulation)”content=”2022年2月23日、FDAは現行のQSR(品質システム規制:21CFR Part 820 Quality System Regulations)をISO 13485:2016に整合させる改正案を公表しました。
QSRは1996年10月7日に現行の規則が発効されて以降、20年以上に渡り米国における医療機器品質システム規制として機能してきたものです。
これまでの間、QSRは軽微な修正はあったものの、改定は一度も実施されてきませんでした。
FDAがQSRによる規制を実施する一方で、品質システム規制に対する当局の期待は時代とともに変化し続けてきました。
日本や欧州、カナダといった多くの国の規制当局は、医療機器の品質システムに関するマネジメント規格であるISO 13485を品質システム規制として取り入れています。
FDAも、他国の規制当局との調和の利点を認識しており、MDSAPへの参加等、規制のハーモナイゼーション活動に取り組んできました。
そして今般、医療機器品質システム規制の国際整合を目指し、QSRのISO13485:2016への調和を目指すこととなりました。
QSRをISO 13485:2016と整合させる規制は「Quality Management System Regulations / QMSR」(品質マネジメントシステム規制)と呼ばれます。
QMSRはISO 13485:2016の要求事項を組み込むことにより、ISO 13485:2016と多くの要求を整合さる予定です。
その一方で、苦情ファイル等の記録の保持に関してISO 13485:2016に追加的要求事項を上乗せしています。
本セミナーでは、QMSRにおける改正点のみではなく、QMSR全般について詳しくわかりやすく解説します。
また、QMSRの要求事項およびISO 13485:2016との差異、QMSRの要求事項を満たすQMS構築について解説します。”]
QSRは1996年10月7日に現行の規則が発効されて以降、20年以上に渡り米国における医療機器品質システム規制として機能してきたものです。
これまでの間、QSRは軽微な修正はあったものの、改定は一度も実施されてきませんでした。
FDAがQSRによる規制を実施する一方で、品質システム規制に対する当局の期待は時代とともに変化し続けてきました。
日本や欧州、カナダといった多くの国の規制当局は、医療機器の品質システムに関するマネジメント規格であるISO 13485を品質システム規制として取り入れています。
FDAも、他国の規制当局との調和の利点を認識しており、MDSAPへの参加等、規制のハーモナイゼーション活動に取り組んできました。
そして今般、医療機器品質システム規制の国際整合を目指し、QSRのISO13485:2016への調和を目指すこととなりました。
QSRをISO 13485:2016と整合させる規制は「Quality Management System Regulations / QMSR」(品質マネジメントシステム規制)と呼ばれます。
QMSRはISO 13485:2016の要求事項を組み込むことにより、ISO 13485:2016と多くの要求を整合さる予定です。
その一方で、苦情ファイル等の記録の保持に関してISO 13485:2016に追加的要求事項を上乗せしています。
本セミナーでは、QMSRにおける改正点のみではなく、QMSR全般について詳しくわかりやすく解説します。
また、QMSRの要求事項およびISO 13485:2016との差異、QMSRの要求事項を満たすQMS構築について解説します。”]
content=”ISO-13485:2016に沿った形の品質マニュアル(ひな形)です。
多くの医療機器企業では、ISO-9001やISO-13485に沿った品質マニュアルを作成していますが、ISO-13485認証審査対応のためには、QSR(品質システム規則:21 CFR 820)に従った品質マニュアルを作成し、説明しなければなりません。
これから作成する医療機器企業様およびISO-13485認証審査を予定している企業様、認証機関から改善指示を受けた企業様向けに、サンプルをご用意いたしました。
MS-Word形式ですので、貴社でご自由に加筆・修正を行っていただけます。”]
Related Articles
- From QSR to QMSR: The FDA’s Historic Transformation of Medical Device Quality System Regulation
- From QSR to QMSR
- The Evolution of Electronic Records Regulations: From FDA Part 11’s Tumultuous Past to Today’s Global Harmonization
- Attending an FDA Inspection: Lessons from a Zero-Observation Success
- Reflections on the Revised GMP Ministerial Ordinance: From Standards to Professional Practice
- From CSV to CSA: A Paradigm Shift in Computer System Validation
{kind=link}
Comment