Understanding the Difference Between “Design” and “Design Control” in Medical Devices

Throughout my consulting work, I have observed that many professionals misunderstand the concept of design control in medical device development.
Throughout my consulting work, I have observed that many professionals misunderstand the concept of design control in medical device development….
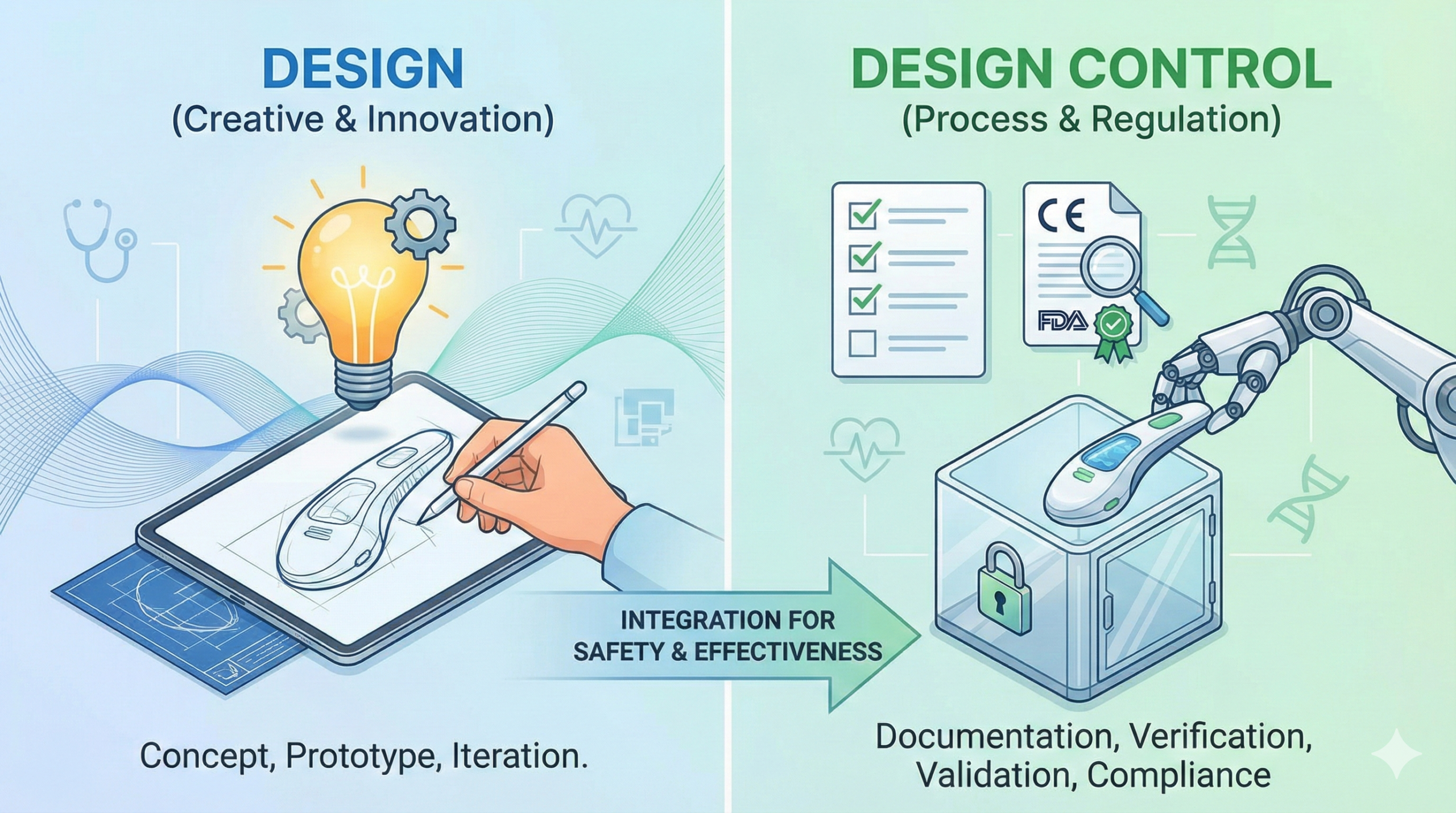
The regulatory requirements and international standards (ISO 13485) do not demand excellence in ‘Design’ per se, but rather excellence in ‘Design Control.’ This distinction is fundamental to achieving regulatory compliance and product success.
Defining “Design” and “Design Control”
Design (in its pure engineering sense) refers to the process of determining the shape, dimensions, functionality, and performance specifications of a medical device, and then documenting these in drawings and specifications. It is an engineering creative activity that reflects each company’s technical capabilities and proprietary knowledge. Following company-specific methodologies, design teams produce drawings and specification documents according to their established practices and expertise.
In contrast, Design Control is a management activity that ensures the design process itself is conducted in compliance with regulatory requirements and a formal Quality Management System (QMS). ISO 13485, Section 7.3, stipulates that “organizations shall plan and carry out design and development in a manner that is appropriate to the subsequent production of medical devices.” Design Control encompasses design reviews (DR), design verification, design validation, risk management integration, and comprehensive documentation requirements.
In practicing Design Control, organizations must comply with their QMS (including conducting design reviews), maintain traceability to intended use and user needs, document design inputs and outputs, perform design verification and validation, manage design changes, and preserve all relevant documents and records in a Design History File (DHF).
The Concept of Files and Practical Implementation
ISO 13485 and other international standards require the creation and maintenance of various “files.” Common examples include the Design History File (DHF), Risk Management File (RMF), and Usability File.
The primary purpose of these files is to organize related documents and records in a manner that facilitates auditing by third-party notified bodies and regulatory authorities. Without such organization, identifying specific documents from the vast collection of design documentation becomes time-consuming and inefficient.
However, the DHF, RMF, and Usability File necessarily overlap in their content. For instance, usability validation records may be included in the DHF, and risk analysis results are relevant to both the DHF and RMF. This overlap reflects the inherent complexity of medical devices and the interconnected nature of design, risk management, and usability considerations.
In practice, organizations address this overlap by creating an Index for each file and developing a Traceability Matrix (Mapping Table) that identifies which sections of which documents satisfy the requirements of each file. This approach enables auditors to quickly locate relevant evidence without requiring the creation of duplicate documentation. It is important to emphasize that one does not create new records entitled “DHF” or “RMF”—rather, one organizes existing documents and makes their relationships explicit through systematic traceability mapping. The file system should be viewed as a tool for organization and validation, not as a burden requiring redundant documentation.
Practical Approaches to File Organization
Many organizations implement file management through a unified document management system combined with traceability matrices. Rather than maintaining physically separate files, this approach uses electronic systems to organize documents while providing clear mapping to the requirements of each regulatory file. This strategy maintains documentation efficiency while fulfilling regulatory compliance requirements.
The Dual Goals of Design in Medical Device Development
In medical device development, design serves two fundamental and equally important objectives.
Goal 1: Creating Regulatory Submission Materials
To bring a medical device to market, regulatory approval from competent authorities (FDA, PMDA, MHRA, European Commission, etc.) is mandatory. From the design phase onward, design teams must consciously collect and organize the data and evidence necessary for regulatory submissions—whether FDA submissions, PMDA approval applications, or EU Technical Documentation.
A common mistake occurs when design engineers focus solely on the engineering aspects of product development without understanding the regulatory pathway and submission requirements. When the regulatory submission phase arrives, it becomes too late to retroactively align the design documentation with the regulatory strategy. Frequently, insufficient verification and validation (V&V) data means that required submission documents (such as FDA Summaries of Safety and Effectiveness [SSED] or EU performance evaluation reports) cannot be completed, necessitating additional testing and studies. Such post-hoc activities extend timelines, increase costs, and risk regulatory deficiency letters.
To avoid these pitfalls, regulatory strategy must be communicated early in the design phase, and design teams must understand the structure and content requirements of the regulatory submission they are building evidence for. Close coordination between regulatory and design functions is essential.
Goal 2: Enabling Manufacturing
A design is not complete simply when the engineering drawings are finalized. Rather, design completion is confirmed only when it has been demonstrated that the design can be consistently manufactured at production volumes, maintain required quality attributes, and achieve acceptable manufacturing costs. If a device cannot be reliably produced at scale with acceptable quality and cost, the design process remains incomplete.
As designs are refined and moved into manufacturing, changes to the design are frequently necessary to address manufacturing feasibility. Conversely, manufacturing challenges often necessitate design modifications. Consequently, many apparent “design changes” are more accurately characterized as manufacturing process adjustments driven by production realities. The iterative interaction between design and manufacturing is a natural and important part of device development.
Ensuring Both Goals Are Met: Interdepartmental Collaboration
Design engineers must fully understand and plan for both objectives from the outset. Several practices support success:
First, regulatory strategy and submission documentation requirements should be discussed early with the design team. Cross-functional communication between regulatory affairs and product development ensures that V&V studies generate the evidence needed for regulatory submissions, avoiding costly redundant testing later.
Second, manufacturing engineering should be integrated into design discussions from the early phases. Adopting Design for Manufacturing (DFM) principles, where manufacturability is considered during design development rather than after design completion, prevents expensive design iterations caused by manufacturing constraints.
Third, key project timelines should be established early and shared across all functions: regulatory submission timelines, manufacturing transition schedules, design freeze dates, and other critical milestones. Clear alignment on these dates reduces misunderstandings and rework.
Recent Regulatory Landscape and Evolving Requirements
The regulatory environment for medical devices has undergone significant changes in recent years, and design control practices must evolve accordingly.
Maturation of EU MDR/IVDR and Increasing Quality Documentation Requirements
The EU Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR) have moved through their transition periods, and competent authorities now enforce full compliance with technical documentation requirements. The Technical Documentation must be more comprehensive and detailed than under the previous Directive regime. Additionally, EU GMP Annex 22, addressing artificial intelligence and machine learning in manufacturing processes, was updated in 2024, introducing new considerations for design control when AI is involved in manufacturing or device functionality.
FDA Computer Software Assurance Guidance Update (2024)
The FDA issued updated guidance on computer software assurance, addressing cybersecurity and software validation for medical devices. Design control processes for software-containing or software-enabled devices must now explicitly address cybersecurity risk management and software assurance methodologies from the design phase onward.
UK MHRA Regulatory Developments Post-Brexit
Following the UK’s exit from the EU, the MHRA has established an independent regulatory framework. Organizations seeking to place devices on the UK market must understand that requirements may differ from EU MDR requirements. Early identification of market-specific regulatory requirements is essential during the technical documentation preparation phase.
Artificial Intelligence and Machine Learning in Medical Devices
Design control processes for devices incorporating AI or machine learning must address AI-specific risks, algorithm verification and validation, model performance monitoring, and strategies for managing continuous learning or model updates. These requirements differ substantially from traditional fixed-design-state approaches and represent a significant evolution in how design control must be implemented.
Conclusion
The distinction between “Design” and “Design Control” in medical device development is not merely semantic—it reflects fundamentally different ways of thinking about product development and regulatory compliance. While engineering creativity and innovation remain central to design excellence, that creativity must be supported by systematic documentation, traceability, and management processes rooted in a robust Quality Management System. Success in the global medical device market depends on design engineers, regulatory affairs professionals, and manufacturing teams working together to achieve both goals: producing evidence-based regulatory submissions and ensuring the designed device can be reliably manufactured. This integrated approach to design control is increasingly recognized as a critical competitive advantage in an evolving regulatory landscape.
Related Articles
- The Importance of Design Control in Medical Devices
- Differences Between Pharmaceutical and Medical Device Regulations Regarding Engineering Design
- Safe Design of Medical Devices: Understanding Risk Management Fundamentals
- Why Should We Focus on Differences from Predicated Devices in Medical Device Design?
- Differences Between Pharmaceuticals and Medical Devices
- The Differences Between Design Verification and Design Review
{kind=link}