
Why does FDA require DHF creation
Regarding DHF, last month also explained, but I would like to address it again as there are many misconceptions.
The Design History File (DHF) corresponds to what is called a design and development file in ISO 13485:2016.
However, there is no requirement by the FDA for the location or configuration of the DHF in the first place.
Common mistakes regarding DHF
In the author’s many years of consulting, he often experiences the following misconceptions about the DHF
- DHF prepares to show to regulatory inspectors
- If the DHF is not created, it will be pointed out
- The DHF maintains a revision history of design and development documents
- Register only the latest version of each design document (technical document)
- Register each design document (technical document) individually
- Manage the number of versions (history) for each design document (technical document)
- Draft versions of each design document (technical document) will also be registered.
- Physically bind all documents in a binder
- Must always be created for each product
These are all mistakes.
A folder should contain a collection of design and development documents.
The DHF does not manage the history of individual documents, but rather the history of design development (documents created at each stage).
We can create a folder for each stage (e.g., design input stage, design output stage) and store the design documents.
DHF shall be created at each stage of the design process by collecting deliverables in the appropriate folder.
DHFs must be created for each individual product (family), and the files to be stored differ for each product.
Why does the FDArequire the creation of a DHFWhy does the FDA require the creation of a DHF?
So why does the FDA require the creation of a DHF?
This is due to the following reasons (See See Design Controls Guide)
- To manage design history
First, the DHF should make it easier for manufacturers and regulators to access the information they need, when they need it, against the scattered design history documents.
Design-related documents are often bound in binders for each department. For example, quality system planning is handled by the QA department, design development by the chief engineer, and so on.
In addition, the design history should not be scattered in various places, such as personal computers, memos, and e-mails. - To enable investigation of complaints (including malfunctions and repairs) with appropriate design documentation (including manufacturing specifications) in the event of a complaint.
A mass of design documents and manufacturing specifications at the date of manufacture must be instantly retrievable. For example, five years ago, three years ago, and last month, the design and manufacturing specifications are different.
A DHF is a collection of the latest design documents (technical documents) at a given point in time for each product (family). In other words, it is like taking a snapshot photo at a certain point in time. Note that this is not filing by individual design document. - To certify that design development was performed in accordance with the design plan. To prevent design development documents from being lost. All design history documents are the property of the manufacturer and not of employees or contractors.
- To improve the following points
The notebooks in the lab are not private property, but the property of the company.
As a result of the engineer’s active participation in the project, separate notebooks will be maintained on each project and submitted to the engineering librarian.
The lab notebooks would be turned over to the employee if he/she leaves the firm.
Product development managers review employee lab notebooks to ensure that records are complete, accurate, and legible on a regular basis.
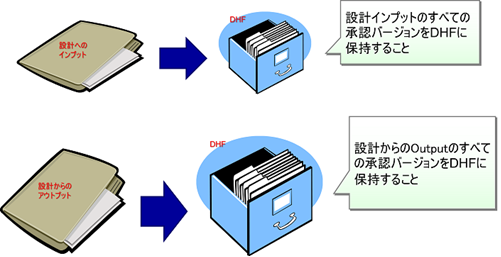
DHF is an index (inventory)
The DHF does not need to be physically bound in a binder, but only needs to be maintained as an index (inventory) of the location of each design document (technical document).
If the design document is an electronic fill, it should be able to be referenced by hyperlink, and if it is a paper document, the storage location (reference point), such as a stack or cabinet location, should be recorded.
This is because it is important that the necessary design documents can be located quickly.
When to register with DHF
The DHF will store all documents related to design controls in a cohesive manner that is approved by the Design Review (DR). The timing of the Design Review (DR) should be considered.
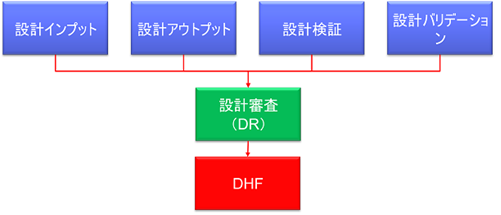
What documents are required by the FDA to be maintained in the DHF?
In 21 CFR 820.30, FDA requires the following three types of records to be maintained in the DHF
820.30(e)Design Review (DR)
820.30(f)design verification
820.30(g)Design Validation
Approximately the following documents will be registered with the DHF
A detailed design development plan that identifies design tasks and deliverables
Approved design inputs and design outputs
Design Review Documentation
Validation Documentation
Controlled design documents and change control records, where appropriate
In other words, all records related to the design as defined in the plan must be registered in the DHF. This includes the design inputs, outputs, and deliverables at each point in the Design Review, Verification, and Validation process.
{kind=link}
Comment