
…
Understanding Validation in Computerized Systems

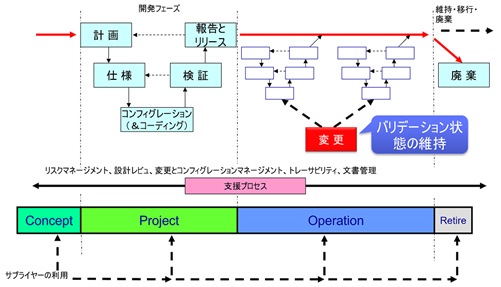
In computerized system validation (CSV) and the modern Computer Software Assurance (CSA) approach, the most important aspect during the operational phase is to maintain the validation status. But what does this actually mean?
To begin with, validation of a computerized system means “ensuring that the system specifications match the user requirements.” During the project phase, the supplier or internal development team defines the requirements, specifies the system, builds it, and tests it to meet user requirements. In other words, at the end of the project phase, the computerized system specifications align with the user’s requirements, and the system is confirmed to be fit for its intended use through documented evidence.
However, user requirements may change during the operational phase after the system is released and begins operation. This is where the challenge of maintaining validation status becomes critical.
Why User Requirements Change During the Operational Phase
User requirements are not static. Even when the system itself remains unchanged, the context in which it operates can evolve significantly. Understanding why and how user requirements change is essential to maintaining validation status effectively.
Common Causes of User Requirement Changes
The following are typical situations where user requirements may change during the operational phase:
1. Changes in Regulatory Requirements
Regulatory authorities continuously update their requirements based on scientific advances, emerging risks, and lessons learned from inspections and adverse events. When regulations change, systems must adapt to meet the new requirements.
Examples include:
- New FDA guidance documents (such as the Computer Software Assurance guidance finalized in September 2025)
- Revisions to EU GMP annexes (e.g., Annex 11 on Computerised Systems, Annex 15 on Qualification and Validation)
- Updates to international standards like ICH guidelines (Q9 on Quality Risk Management, Q10 on Pharmaceutical Quality System, Q12 on Lifecycle Management)
- Changes in national regulations or pharmacopeial requirements
These regulatory changes may necessitate new system functionalities, enhanced audit trails, additional data integrity controls, or modified validation documentation approaches. For instance, the shift from traditional CSV to risk-based CSA approaches means organizations must reassess their validation strategies and potentially enhance their systems to support continuous assurance activities.
2. Changes in the Interpretation of Regulatory Requirements
Sometimes, regulatory requirements themselves do not change, but the interpretation or understanding of those requirements evolves. This often occurs when:
- Regulatory inspections identify gaps or areas for improvement
- Internal audits reveal deficiencies in current practices
- Industry guidance documents provide new interpretations
- Warning letters to other companies clarify regulatory expectations
- New court rulings or legal interpretations affect compliance strategies
For example, if a Quality Management System (QMS) receives observations during a regulatory inspection regarding data integrity practices, procedures and system configurations may need to be revised to address the findings—even though the underlying regulations (such as 21 CFR Part 11 for electronic records) have not changed. This, in turn, may alter the user requirements for computerized systems supporting those procedures, potentially requiring enhanced audit trail functionality, stricter access controls, or additional data backup procedures.
3. Organizational Changes
Companies undergo various organizational transformations that can impact system requirements:
- Mergers and acquisitions requiring system integration or data migration
- Departmental restructuring changing workflows and approval hierarchies
- Implementation of new quality management systems or enterprise resource planning (ERP) systems
- Changes in business processes or manufacturing strategies
- Addition or removal of product lines or therapeutic areas
- Changes in personnel or organizational roles affecting system access and permissions
These organizational changes often introduce new users, modify data flows, alter reporting structures, or create new integration requirements—all of which can change what is required from the computerized system. For instance, a merger might require two previously separate laboratory information management systems (LIMS) to share data, necessitating new interfaces and modified validation scopes.
4. Product Changes
Changes to the products manufactured or tested can significantly impact system requirements:
- Introduction of new products with different specifications or testing requirements
- Formulation changes requiring different analytical methods
- Manufacturing process modifications affecting in-process controls
- Changes to critical quality attributes (CQAs) or critical process parameters (CPPs)
- Scale-up or scale-down of manufacturing operations
- Transfer of products between manufacturing sites
Product changes can alter the risk profile associated with system failures. For instance, if a company begins manufacturing a higher-risk product (such as a sterile injectable or an oncology drug instead of an oral solid dosage form), the criticality of systems supporting that product increases. A system function that was previously classified as low-risk or “not high process risk” (in CSA terminology) may become high-risk, requiring more rigorous validation, additional controls, and enhanced assurance activities.
5. Changes in Procedures
Standard Operating Procedures (SOPs) and work instructions are living documents that are regularly updated to reflect best practices, lessons learned, and continuous improvement initiatives:
- Revision of SOPs based on CAPA (Corrective and Preventive Action) outcomes
- Updates to work instructions following process improvements or lean manufacturing initiatives
- Changes to quality agreements with suppliers or contract manufacturing organizations
- Modifications to training programs or competency requirements
- Implementation of new quality initiatives, such as enhanced data integrity programs
- Updates to validation or change control procedures themselves
When procedures change, the way users interact with computerized systems may also change. New data capture requirements, modified workflows, additional approval steps, or different reporting needs may emerge, effectively changing the user requirements for the system. For example, if an SOP is revised to require electronic signatures at additional approval points, the system must be validated to ensure those signature requirements are properly enforced.
The Critical Link Between Changes and Risk
A fundamental principle in modern validation approaches, as outlined in ICH Q9(R1) Quality Risk Management and GAMP 5 Second Edition, is that validation and assurance activities should be risk-based. This means:
Validation activities should be scaled based on:
- The intended use of the system or specific software features
- The process risk (the risk that a system failure could adversely impact product quality, patient safety, or data integrity)
- The severity of potential consequences if the system fails to perform as intended
- The likelihood of such failures occurring
When user requirements change due to any of the five factors above, the risk profile may also change:
- A product change may increase the severity of a potential system failure
- A regulatory requirement change may identify new hazards that were not previously considered
- An organizational change may affect the detectability of errors or the effectiveness of existing controls
- A procedure change may alter the criticality of certain system functions
If the risk changes, the validation strategy must be reassessed. For example:
- A Laboratory Information Management System (LIMS) used only for non-GMP research might later be used for GMP stability testing. The system hasn’t changed, but its intended use and associated risks have increased dramatically, requiring enhanced validation.
- A manufacturing execution system (MES) originally validated for a low-risk over-the-counter product might later be used for a critical life-saving drug. The system functionality remains the same, but the potential patient impact is much greater, necessitating additional controls and more comprehensive validation.
This is why maintaining validation status is not simply about documenting that nothing has changed. It requires actively monitoring for changes in context, requirements, and risk, and then taking appropriate action to ensure the system continues to meet its intended use.
What is “Maintaining Validation Status”?
Now that we understand why user requirements change, we can define what it means to maintain validation status:
Maintaining validation status means ensuring that, throughout the operational phase, the computerized system specifications continue to meet current user requirements, and that the system consistently performs as intended under actual operating conditions.
This is achieved through a systematic change control process that evaluates, approves, implements, and verifies all changes that could impact the validated state of the system. The goal is to ensure that the original validated state—where system specifications matched user requirements—remains true even as those user requirements evolve.
Change Control: The Mechanism for Maintaining Validation Status
Change control is the primary tool for maintaining validation status during the operational phase. As defined in EU GMP Annex 15:
“A formal change control system should be in place to plan, evaluate and document all changes that may affect the validated status of facilities, systems, equipment or processes. The intent is to determine the need for action that would ensure and document that the system is maintained in a validated state.”
A robust change control system for computerized systems includes the following key elements:
1. Change Identification and Documentation
- Clear identification of what is changing (system, procedure, product, organization, or regulation)
- Formal documentation in a change control record
- Assignment of a unique change control number for traceability
2. Impact Assessment
- Evaluation of how the change affects the computerized system
- Assessment of whether the change alters user requirements
- Determination of whether the system specifications still meet the (potentially changed) user requirements
- Risk assessment following ICH Q9 principles
3. Determination of Action Required
- Based on the impact assessment, determine what actions are needed:
- No action required (change does not affect the system)
- Documentation update only
- Minor validation activities (targeted testing)
- Partial revalidation (specific modules or functions)
- Full revalidation (entire system)
- The level of validation activity should be proportional to the risk
4. Review and Approval
- Cross-functional review by subject matter experts
- Quality Assurance approval (mandatory)
- User and IT stakeholder approval as appropriate
- Risk-based decision making
5. Implementation
- Execution of approved changes in a controlled manner
- Following established procedures and work instructions
- Proper version control and configuration management
6. Verification and Testing
- Confirmation that changes were implemented correctly
- Testing appropriate to the risk level (from simple verification to comprehensive validation protocols)
- Regression testing to ensure existing functionality is not adversely affected
- Use of risk-based testing strategies (scripted, unscripted, automated, or combinations)
7. Documentation and Closure
- Completion of all required documentation
- Update of validation master plans, validation reports, or assurance documentation
- Revision of SOPs and training materials if needed
- Formal closure of the change control record
Examples of When Change Control is Needed
Example 1: New Regulatory Requirement A new FDA guidance emphasizes the importance of electronic batch record review completeness. Your Manufacturing Execution System (MES) currently allows batch record approval even if certain optional fields are left blank. User requirements now change to mandate that all critical fields must be completed before approval.
Action Required: Change control to modify the MES to enforce mandatory field completion. Impact assessment determines this is a medium-risk change affecting batch release. Validation activities include functional testing of the new field validation logic and regression testing of the approval workflow.
Example 2: Product Change Your company introduces a new parenteral (injectable) product line in addition to existing oral solid dose products. The same Enterprise Resource Planning (ERP) system will be used for inventory and production management. However, the consequences of inventory errors are now more severe given the higher risk of the new product.
Action Required: Change control to reassess the ERP system validation. User requirements haven’t explicitly changed, but the risk profile has increased. Impact assessment determines that functions previously validated at a basic level (like lot number tracking) now require enhanced validation. Additional validation activities focus on traceability and error prevention.
Example 3: Organizational Change After a corporate merger, two quality control laboratories will share the same Laboratory Information Management System (LIMS). New workflows are needed to accommodate users from both legacy organizations, and data integration requirements emerge.
Action Required: Change control to implement new LIMS configurations for merged operations. Impact assessment identifies changes to user roles, permissions, workflows, and interfaces. Validation activities include testing new workflows, verifying data migration integrity, and confirming appropriate access controls for all user groups.
Example 4: Procedure Change A CAPA investigation identifies that sample tracking procedures need to be enhanced. The SOP is revised to require scanning sample barcodes at two additional checkpoints in the laboratory workflow. The LIMS must now capture data at these new checkpoints.
Action Required: Change control to configure LIMS for additional tracking points. Impact assessment determines this is a low-to-medium risk change that enhances traceability. Validation activities include testing the new scanning workflows and confirming data is correctly recorded in the system.
Example 5: System Vendor Update The vendor of your validated Electronic Document Management System (EDMS) releases a mandatory security patch to address a critical vulnerability. The patch also includes minor user interface changes.
Action Required: Change control to evaluate and implement the vendor update. Impact assessment reviews vendor documentation and determines that security patches are critical but UI changes are cosmetic. Risk-based validation includes verification that core document control functions (creation, review, approval, archiving) still work correctly. Regression testing focuses on high-risk functions, while lower-risk cosmetic changes receive lighter verification.
The Relationship with Risk-Based Approaches
Modern validation approaches, as embodied in the FDA’s Computer Software Assurance guidance (2025) and GAMP 5 Second Edition (2022), emphasize risk-based strategies. This has important implications for maintaining validation status:
Traditional CSV Approach
In traditional CSV, maintaining validation status often meant:
- Periodic revalidation at fixed intervals (e.g., every 3 years)
- Extensive documentation for all changes, regardless of risk
- Full regression testing for even minor modifications
- Heavy reliance on scripted Installation Qualification/Operational Qualification/Performance Qualification (IQ/OQ/PQ) protocols
Modern CSA/Risk-Based Approach
Under CSA and risk-based validation principles:
- Continuous assurance rather than periodic revalidation
- Proportional validation activities based on risk assessment
- Leverage of system-generated evidence (audit trails, logs, monitoring data)
- Flexible testing approaches (scripted, unscripted, exploratory, automated)
- Vendor documentation utilization to reduce duplication
However, the fundamental requirement remains the same: the system must maintain a validated state where specifications continue to meet user requirements. The difference is in how we demonstrate and document this maintained state.
Applying Risk-Based Thinking to Change Control
When evaluating changes through the change control process, apply ICH Q9 risk management principles:
Risk Assessment Questions:
- What is the intended use of the system or affected feature?
- What is the process risk? (Could a failure impact product quality, patient safety, or data integrity?)
- What is the severity of the potential impact?
- What is the likelihood of failure?
- What controls are already in place?
- Are additional controls or validation activities needed?
Risk-Based Decision Making:
- High process risk functions (e.g., batch release decisions, critical test results, electronic signatures on GMP records): Require rigorous assurance activities, comprehensive testing, and formal documentation
- Not high process risk functions (e.g., optional report formatting, non-GMP dashboards): May use streamlined approaches, unscripted testing, or rely on vendor documentation
- Medium risk functions: Apply judgment and scale activities appropriately
Example Risk Classification: Consider a Laboratory Information Management System (LIMS):
- High process risk: Sample test result entry and approval (directly impacts batch release decisions)
- Medium process risk: Instrument integration and data transfer (important for efficiency and accuracy, but subject to analyst review)
- Not high process risk: Dashboard color schemes or user preference settings (no GMP impact)
When changes occur, the validation activities required should reflect these risk levels. A change to the result approval workflow requires comprehensive validation, while a dashboard color change might require only basic verification that the change was correctly implemented.
Regulatory Expectations for Maintaining Validation Status
Regulatory authorities clearly expect organizations to maintain validation status throughout the system lifecycle:
FDA 21 CFR 820.70(i) (Medical Device Quality System Regulation): “When computers or automated data processing systems are used as part of production or the quality system, the manufacturer shall validate computer software for its intended use according to an established protocol. All software changes shall be validated before approval and issuance.”
FDA 21 CFR Part 211.68 (Pharmaceutical cGMP): “Appropriate controls shall be exercised over computer or related systems to assure that changes in master production and control records or other records are instituted only by authorized personnel.”
EU GMP Annex 11: “The system should be periodically evaluated to confirm that it remains in a validated state and is compliant with relevant requirements… Change control procedures should be in place.”
EU GMP Annex 15: “Qualification and validation should not be considered as one-off exercises. A formal change control system should be in place… Changes to facilities, supporting systems, equipment and processes which may affect product quality should be formally documented, risk assessed and approved before implementation.”
ICH Q10 (Pharmaceutical Quality System): “Change management systems should ensure that changes are proposed, evaluated for potential impact, approved prior to implementation, implemented in a controlled manner, and reviewed for effectiveness.”
The upcoming Quality Management System Regulation (QMSR), which harmonizes FDA 21 CFR Part 820 with ISO 13485:2016 and takes effect February 2, 2026, further emphasizes lifecycle management and change control as essential QMS elements.
Common regulatory inspection findings related to maintenance of validation status include:
- Inadequate change control procedures or failure to follow them
- Changes implemented without proper impact assessment
- Lack of revalidation when required
- Insufficient documentation of change rationale and validation activities
- Cumulative effect of multiple small changes not assessed
- Systems not periodically reviewed to confirm maintained validated state
Practical Steps for Maintaining Validation Status
To effectively maintain validation status during the operational phase, organizations should implement the following practices:
1. Establish a Robust Change Control System
- Develop and document change control procedures applicable to all GXP computerized systems
- Define clear triggers for when change control is required
- Establish a cross-functional change control committee or review board
- Integrate computerized system change control with the site-wide change control process
- Ensure change control procedures are risk-based and appropriately scaled
2. Train Personnel
- Ensure all stakeholders understand the importance of maintaining validation status
- Train users to recognize when changes might affect validation
- Train validators and quality assurance personnel on risk assessment and appropriate validation responses
- Emphasize that maintaining validation status is everyone’s responsibility
3. Monitor for Changes
- Establish mechanisms to detect changes that might affect user requirements:
- Regular review of regulatory updates
- Monitoring of inspection trends and warning letters
- Internal audit programs
- Product change management processes
- Organizational change communications
- Subscribe to regulatory news services and industry association updates
- Participate in industry working groups to stay informed of evolving expectations
4. Conduct Impact Assessments
- For each identified change, systematically assess impact on computerized systems
- Use structured risk assessment tools (e.g., FMEA, risk matrices)
- Document the rationale for decisions (especially when determining that no action is needed)
- Involve appropriate subject matter experts in assessments
5. Execute Validation Activities Proportional to Risk
- Apply risk-based principles to determine the appropriate level of validation
- For high-risk changes, conduct comprehensive validation including formal protocols
- For medium-risk changes, perform targeted testing and verification
- For low-risk changes, rely on basic verification and documentation updates
- Use a combination of scripted, unscripted, and automated testing as appropriate
- Leverage vendor documentation where justified
6. Document Thoroughly
- Maintain complete change control records
- Document the rationale for all decisions, especially risk-based decisions
- Keep validation documentation current (update validation master plans, summary reports)
- Ensure traceability between changes, impact assessments, and validation activities
- Use electronic quality management systems (eQMS) to facilitate documentation and trending
7. Perform Periodic Reviews
- Schedule periodic reviews of validated systems (e.g., annually)
- Review change control history to identify trends
- Assess cumulative impact of multiple changes
- Verify that validation documentation remains current and accurate
- Confirm that the system continues to meet its intended use
- Update risk assessments based on operational experience
8. Integrate with Other Quality Systems
- Link change control to CAPA processes
- Connect validation activities to quality risk management
- Coordinate with deviation management and investigations
- Align with product lifecycle management (ICH Q12)
- Support continuous improvement initiatives
Conclusion
Maintaining validation status is a critical ongoing activity during the operational phase of computerized systems. It ensures that as the world changes around the system—through new regulations, organizational evolution, product changes, or procedural updates—the system continues to meet its intended use and performs reliably.
The key to maintaining validation status is recognizing that user requirements are dynamic, not static. Through systematic change control, risk-based impact assessments, and appropriate validation activities, organizations can ensure that system specifications always align with current user requirements.
While modern approaches like Computer Software Assurance (CSA) and GAMP 5 Second Edition offer more flexible, risk-based strategies, the fundamental principle remains unchanged: validated systems must remain validated. Change control is the mechanism that makes this possible, bridging the gap between initial validation and ongoing assurance throughout the system lifecycle.
By understanding the common causes of user requirement changes, implementing robust change control processes, applying risk-based thinking, and maintaining appropriate documentation, organizations can successfully maintain validation status, meet regulatory expectations, and ultimately ensure product quality and patient safety.
Key Takeaways
Documentation is essential but should be appropriate to risk – Record decisions, rationale, and evidence proportional to the risk level
Validation means system specifications match user requirements – This alignment must be maintained throughout the operational phase
User requirements change for five main reasons – Regulatory changes, interpretation changes, organizational changes, product changes, and procedure changes
Changes in requirements often mean changes in risk – Risk reassessment is essential when context changes
Change control is the mechanism for maintaining validation status – A systematic process evaluates, approves, and verifies changes
Modern approaches emphasize risk-based activities – CSA and GAMP 5 Second Edition provide flexible, proportional strategies
Regulatory expectations are clear – Authorities require maintained validation throughout the system lifecycle
Periodic review supplements change-driven activities – Regular reviews catch cumulative effects and confirm continued suitability
Related products
[blogcard url=https://ecompliance.jp/qms-md/ title=”QMS(手順書)ひな形 医療機器関連” ] [blogcard url=https://ecompliance.jp/qms-rx/ title=”QMS(手順書)ひな形 医薬品関連” ] [blogcard url= https://ecompliance.co.jp/SHOP/CSV-5KY-LIVE-000010.html title=”【VOD】CSVセミナー【第10講】”] [blogcard url= https://ecompliance.co.jp/SHOP/L_MDCSV.html title=”【VOD】医療機器企業におけるCSV実践セミナー“] [blogcard url= https://ecompliance.co.jp/SHOP/BOOK-CSV2.html title=”【書籍】【超入門】コンピュータ化システムバリデーション”] [blogcard url= https://ecompliance.co.jp/SHOP/EL-026.html title=”【セミナービデオ】現場目線で考える!すぐ活用出来る! コンピュータ化システムバリデーション(CSV)超入門2020″]
Related Articles
- What is Validation (Revised Edition)
- What is Process Validation?
- Maintaining Validation State in Computerized System Validation
- What is Process Validation?
- What are the conditions for process validation?
- Understanding the True Purpose of Performance Qualification (PQ) in Computerized System Validation
{kind=link}
Comment