
Why FMEA Should Not Be Used for Medical Device Design

Introduction
Risk management is the most critical process for ensuring patient safety in medical device design and development. While Failure Modes and Effects Analysis (FMEA) has been widely adopted across many industrial sectors, there is a significant movement to apply it to medical device design as well. However, this approach harbors serious problems. This article explains in detail why FMEA is not appropriate for medical device design.
Risk management is the most critical process for ensuring patient safety in medical device design and development. While Failure Modes and Effects Ana…
What is FMEA?
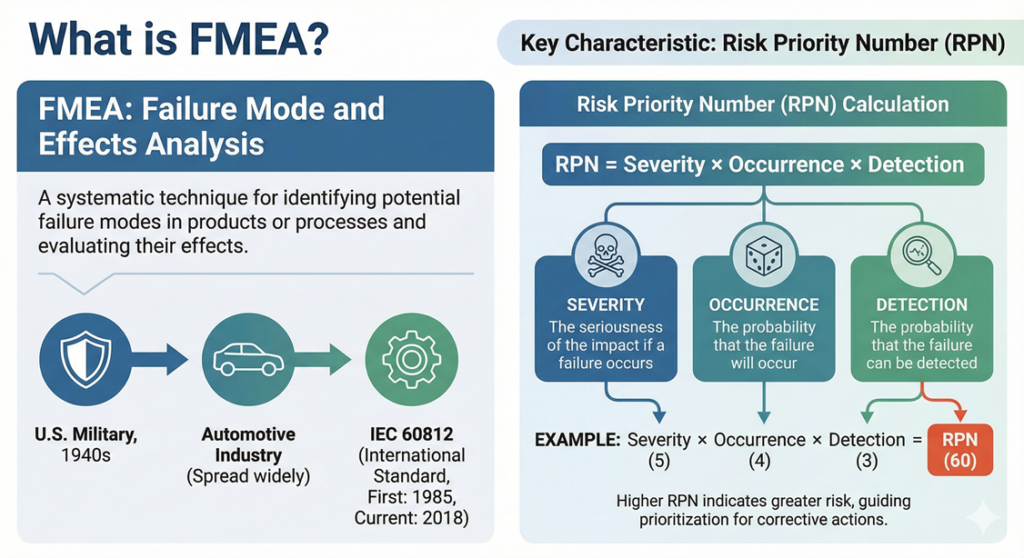
FMEA (Failure Mode and Effects Analysis) is a technique for identifying potential failure modes in products or processes and evaluating their effects. Originally developed by the U.S. military in the 1940s, it subsequently spread widely, particularly throughout the automotive industry. FMEA is now standardized internationally as IEC 60812 (first published in 1985, with the current third edition released in 2018).
The most distinctive characteristic of FMEA is its use of an indicator called the Risk Priority Number (RPN). RPN is calculated as the product of three elements:
- Severity: The seriousness of the impact if a failure occurs
- Occurrence: The probability that the failure will occur
- Detection: The probability that the failure can be detected
For example, if each element is rated on a scale of 1 to 10, then RPN = Severity × Occurrence × Detection, with a maximum value of 1000.
Problems with Medical Device Design
The Pitfall of Detection
The concept of “detection” in FMEA is the single greatest factor that makes it inappropriate for application to medical device design. This element means that if a failure or defect is easy to detect, the risk is evaluated as lower.
However, consider the environment in which medical devices are used. For medical devices used on patients, even if detection is easy, it is too late once serious harm has occurred. For example, even if a ventilator failure can be detected by an alarm, the patient’s life may be in danger at that very moment.
Conflict Between Corporate Profit and Patient Safety
The RPN approach in FMEA is fundamentally a way of thinking that prioritizes corporate economics. When detection is high (easy to detect), the RPN value becomes smaller, and the priority for risk countermeasures decreases. This can lead to judgments such as “even if a problem occurs, we can discover it, so we don’t need to spend money on countermeasures.”
Such thinking cannot be permitted in medical devices. Patient safety must be absolutely guaranteed regardless of ease of detection.
The Importance of ISO 14971
For risk management of medical devices, the international standard ISO 14971 exists. This standard defines risk management requirements specific to medical devices and employs a fundamentally different approach from FMEA.
ISO 14971 evaluates risk using the following two elements:
- Severity of harm: The seriousness of the harm
- Probability of occurrence of harm: The probability that the harm will occur
The concept of “detection” is not included here. This is an intentional design. For medical devices, as long as there is a possibility that harm may occur, risk must be reduced to an acceptable level regardless of whether it can be detected.
The current version is ISO 14971:2019 (third edition), which was released in December 2019 together with the technical guidance document ISO/TR 24971:2020. The 2019 edition represents a significant evolution from previous versions, with enhanced focus on benefit-risk analysis, expanded requirements for production and post-production activities, and stronger alignment with regulatory requirements in major markets worldwide.
Additionally, in 2021, the European harmonized version ISO 14971:2019+A11:2021 was published. The A11 annex describes how the requirements of ISO 14971 can be used to demonstrate conformity with the General Safety and Performance Requirements (GSPR) of the EU Medical Device Regulation (MDR 2017/745) and In Vitro Diagnostic Regulation (IVDR 2017/746).
Principles of Risk Control
ISO 14971 establishes clear priorities for risk control:
- Inherently safe design: Eliminate or reduce risk through design itself
- Protective measures: Protective measures in the medical device itself or in the manufacturing process
- Information for safety: Provision of information about residual risks
This hierarchical approach focuses on reducing the risk itself, not simply detecting risks.
The importance of this hierarchy cannot be overstated. Manufacturers must first attempt to eliminate hazards through design changes. Only when this is not feasible should they move to implementing protective measures such as guards, interlocks, or alarms. Information for safety, such as warnings in user manuals or training, is considered the least effective risk control measure and should only be relied upon when higher-level controls are not practical.
Appropriate Use Cases for FMEA
FMEA is not entirely useless. Even in the medical device industry, it is effective for risk management of manufacturing processes. In manufacturing processes, detection and removal of defective products are important elements of quality control, so it makes sense to consider detection.
However, the design stage is a different matter. Design risks can directly affect patients, so risk assessment that relies on detection is inappropriate.
It is worth noting that FMEA can be used as a complementary tool within a comprehensive ISO 14971-compliant risk management system. For instance, Design FMEA (DFMEA) can help identify component-level failure modes during the early design phase, while Process FMEA (PFMEA) can support manufacturing quality. However, these must be integrated into an overarching risk management framework that does not rely on detection as a primary risk reduction strategy.
Practical Implications
Design engineers and risk management personnel at medical device manufacturers should pay attention to the following points:
1. Correct Understanding of Standards
It is important to accurately understand the requirements of ISO 14971 and clearly recognize the differences from FMEA. Confusing the two not only fails to meet regulatory requirements but can also threaten patient safety.
Manufacturers should also be aware that ISO/TR 24971:2020 provides extensive guidance on applying ISO 14971, including clarifications on benefit-risk considerations, post-production information collection, and the relationship between ISO 14971 and other standards such as IEC 62366-1 (usability engineering) and IEC 60601-1 (electrical safety).
2. Building a Risk Management File
When constructing a Risk Management File based on ISO 14971, it is necessary to conduct risk assessment that does not depend on detection. For each risk, countermeasures through inherently safe design should be given top priority.
The Risk Management File should document the complete risk management process throughout the device lifecycle, including:
- Risk management plan
- Risk analysis (hazard identification and risk estimation)
- Risk evaluation
- Risk control measures and their verification
- Evaluation of overall residual risk
- Production and post-production information
- Risk management report
3. Fostering Organizational Culture
A shift is needed from the mindset of “it’s okay because we can detect it” to “prevent harm from occurring in the first place.” This is not merely a change in methodology but involves transformation of organizational culture.
This cultural transformation requires commitment from top management, appropriate training for all personnel involved in device development, and continuous reinforcement through quality management system processes. Organizations should encourage open discussion of risks and create an environment where safety concerns can be raised without fear of criticism.
Regulatory Authority Perspective
Medical device regulatory authorities around the world require application of ISO 14971. Risk management using FMEA is highly likely to be judged as not meeting regulatory requirements.
For example, the U.S. FDA recognizes ISO 14971 as a consensus standard and expects manufacturers to implement risk management processes consistent with this standard. With the implementation of the Quality Management System Regulation (QMSR) in February 2026, which incorporates ISO 13485:2016, the importance of ISO 14971-compliant risk management will increase further in the U.S. market.
In the European Union, under the MDR and IVDR, manufacturers must demonstrate compliance with General Safety and Performance Requirements, which explicitly reference risk management in accordance with the state of the art. The harmonized European standard EN ISO 14971:2019+A11:2021 provides a presumption of conformity with these requirements.
Similarly, regulatory authorities in other major markets including Health Canada (under the Medical Device Single Audit Program – MDSAP), Australia’s TGA, and Japan’s PMDA all expect compliance with ISO 14971 as part of their quality management system requirements.
Key Differences Between FMEA and ISO 14971
To further clarify why FMEA is unsuitable for medical device design, it is helpful to examine specific differences in a structured manner:
| Aspect | FMEA (IEC 60812) | ISO 14971 |
| Primary Focus | Component/process failure modes | Hazards and hazardous situations |
| Risk Definition | RPN (Severity × Occurrence × Detection) | Probability of occurrence of harm × Severity of harm |
| Detection Consideration | Integral to risk calculation | Not part of risk definition (detection relates to risk control effectiveness) |
| Scope of Analysis | Primarily failure conditions | Both normal use and reasonably foreseeable misuse |
| Severity Basis | System performance impact | Harm to patients, users, or others |
| Starting Point | Components or process steps (bottom-up) | Intended use and hazards (top-down) |
| Timing | Typically later in development | From initial concept through entire lifecycle |
| Risk Control Priority | Not explicitly specified | Clear hierarchy: inherent safety → protective measures → information |
| Benefit Consideration | Not addressed | Explicit benefit-risk analysis required (2019 edition) |
| Post-Market Activities | Not typically included | Comprehensive production and post-production requirements |
This table illustrates that while FMEA can be a valuable reliability engineering tool, it lacks several critical elements required for comprehensive medical device risk management.
Benefit-Risk Considerations
One of the significant enhancements in ISO 14971:2019 is the increased emphasis on benefit-risk analysis. Medical devices, by their nature, carry inherent risks, but these risks must be balanced against the intended medical benefits. ISO 14971:2019 now explicitly requires manufacturers to:
- Define the intended medical benefit of the device
- Ensure that the overall residual risk is acceptable when weighed against the intended medical benefit
- Consider whether the intended benefit can be achieved with lower risk
This benefit-risk framework is entirely absent from traditional FMEA methodology, which focuses purely on failure modes without considering the clinical value that the device provides. For medical devices, especially those used in life-threatening situations or for treating serious conditions, some level of residual risk may be acceptable if the medical benefit sufficiently outweighs that risk.
Regulatory authorities worldwide, including the FDA in its guidance “Factors to Consider Regarding Benefit-Risk in Medical Device Product Availability, Compliance, and Enforcement Decisions,” increasingly emphasize this benefit-risk paradigm in their decision-making processes.
Production and Post-Production Information
ISO 14971:2019 significantly expanded requirements for gathering and analyzing information during production and post-production phases. Manufacturers must establish systematic processes to:
- Collect information from production activities (such as trending of non-conformances and CAPA data)
- Gather post-production information including:
- Customer complaints and Medical Device Reports (MDRs)
- Published literature and competitive analyses
- Field safety corrective actions (recalls)
- User feedback and real-world performance data
This information must be regularly reviewed to determine if:
- Previously unrecognized hazards are present
- Estimated risks arising from a hazard are no longer acceptable
- The overall residual risk is no longer acceptable
If any of these conditions are identified, manufacturers must initiate appropriate risk management activities, which may include design changes, protective measures, or updates to information for safety.
This lifecycle approach to risk management far exceeds the scope of traditional FMEA, which is typically a point-in-time analysis rather than an ongoing process of information gathering and risk reassessment.
The Role of Detection in Risk Control
While ISO 14971 does not include detection as part of the definition of risk, this does not mean that detection is irrelevant. Detection becomes important in the context of risk control measures, particularly:
- Verification of risk control effectiveness: Determining whether implemented risk controls actually achieve the intended risk reduction
- Manufacturing process controls: Ensuring that manufacturing processes consistently produce devices meeting specifications (where Process FMEA can be valuable)
- User interface design: Evaluating whether users can recognize hazardous situations and respond appropriately
However, even in these contexts, detection serves to verify or support risk control measures rather than substitute for inherent safety design. A well-designed medical device should not rely primarily on detection to prevent harm, as this places the burden of safety on users or post-manufacture inspection rather than on robust design.
International Recognition and Harmonization
The global harmonization around ISO 14971 reflects a consensus among international experts and regulatory authorities that medical device risk management requires a specialized approach distinct from general industrial reliability methods like FMEA.
ISO 14971 was developed by ISO Technical Committee 210 (Quality management and corresponding general aspects for medical devices) in collaboration with IEC Subcommittee 62A (Common aspects of electrical equipment used in medical practice). This cross-disciplinary development ensures that the standard addresses the unique challenges of medical devices, including electrical safety, usability, biocompatibility, cybersecurity, and other specialized concerns.
Major quality management and risk-related standards in the medical device field explicitly reference or build upon ISO 14971, including:
- ISO 13485:2016: Quality management systems for medical devices (requires risk-based thinking throughout)
- IEC 62366-1:2015: Application of usability engineering to medical devices (integrates with ISO 14971 for use-related risks)
- ISO 10993-1:2018: Biological evaluation of medical devices (explicitly conducted within an ISO 14971 risk management process)
- IEC 60601-1: Medical electrical equipment safety (references ISO 14971 for risk management)
- ISO 14155: Clinical investigation of medical devices for human subjects (requires risk management per ISO 14971)
This extensive interconnection demonstrates that ISO 14971 is not merely one option among many for medical device risk management, but rather the foundational framework upon which the entire regulatory system is built.
Common Misconceptions About FMEA in Medical Devices
Several misconceptions persist in the medical device industry regarding the use of FMEA:
Misconception 1: “FMEA is required by regulations”
- Reality: FMEA itself is not mandated by any major medical device regulation. ISO 14971 is the required standard for risk management.
Misconception 2: “Using FMEA demonstrates compliance with ISO 14971”
- Reality: FMEA alone does not meet ISO 14971 requirements. At minimum, FMEA would need to be substantially modified (eliminating detection from risk calculation, addressing normal use scenarios, focusing on harm rather than system failure) to begin approaching ISO 14971 requirements.
Misconception 3: “High RPN values identify the most important risks”
- Reality: In medical devices, severity of harm should be the primary driver of risk management activities. A failure mode with catastrophic severity but low occurrence and high detection would have a moderate RPN in traditional FMEA, yet clearly requires aggressive risk control in medical device design.
Misconception 4: “Detection makes risks acceptable”
- Reality: For medical device patient safety, detection of a problem after a device is manufactured and released does not make the underlying hazard acceptable. Inherent safe design is the priority.
Misconception 5: “FMEA has no place in medical device development”
- Reality: While FMEA should not be the primary risk management tool for design, it can serve complementary roles in manufacturing process control and as one technique among others for failure mode identification, provided it is integrated within an ISO 14971-compliant system.
Practical Implementation Guidance
For organizations transitioning from FMEA-based approaches to ISO 14971-compliant risk management, the following steps are recommended:
- Gap Analysis: Compare current risk management practices against ISO 14971:2019 requirements to identify deficiencies
- Training: Ensure all personnel involved in device development understand ISO 14971 principles and the critical differences from FMEA
- Process Revision: Update risk management procedures to align with ISO 14971, including:
- Hazard identification from intended use and reasonably foreseeable misuse
- Risk estimation based solely on severity and probability (without detection)
- Structured risk control following the prescribed hierarchy
- Comprehensive benefit-risk evaluation
- Production and post-production information systems
- Documentation Templates: Revise risk management templates and tools to support ISO 14971 requirements rather than FMEA formats
- Integration: Ensure risk management is integrated throughout the quality management system, design controls, and post-market surveillance processes
- Continuous Improvement: Establish mechanisms for ongoing review and enhancement of risk management practices based on internal audits, management reviews, and regulatory feedback
Organizations should also leverage the extensive guidance in ISO/TR 24971:2020, which provides detailed explanations, examples, and rationale for ISO 14971 requirements.
Conclusion
FMEA is an effective risk analysis method in many industries, but it is not suitable for medical device design. The reason is that the concept of detection leads to thinking that prioritizes corporate economics over patient safety.
For medical device design, risk management based on ISO 14971 should be implemented. This is not merely a response to regulatory requirements but is the result of prioritizing patient safety.
Everyone involved in medical device development has a responsibility to understand these differences and choose appropriate methodologies. In medical devices that hold patients’ lives and health in trust, no compromise is permitted. Choosing and implementing correct risk management methodologies is what leads to the development of safe and effective medical devices.
The medical device industry continues to evolve, with increasing complexity in technologies such as software as a medical device (SaMD), artificial intelligence and machine learning, cybersecurity, and interoperability. ISO 14971 provides a flexible yet robust framework that can address these emerging challenges. By adhering to the principles established in this international standard and avoiding the pitfalls of inappropriate application of FMEA to design, manufacturers can fulfill their primary obligation: ensuring that their devices benefit patients without causing unacceptable harm.
As regulations continue to harmonize globally around ISO 14971 and as enforcement becomes increasingly rigorous, the time for organizations to transition away from FMEA-based design risk management approaches is now. Patient safety depends on it, regulatory compliance requires it, and the medical device community consensus supports it. The path forward is clear: ISO 14971 is the appropriate standard for medical device risk management throughout the entire product lifecycle.
Related Articles
- FMEA should not be used in medical device design
- FMEA Must Not Be Used in Medical Device Design for Risk Evaluation Purposes
- Why Should We Focus on Differences from Predicated Devices in Medical Device Design?
- Can GAMP Be Used in Medical Device Companies?
- Understanding FMEA in Pharmaceutical and Medical Device Industries
- The Importance of Design Control in Medical Devices
{kind=link}